New technique allows tracking of gene expression over generations of cells as they specialize.
By combining sophisticated RNA sequencing technology with a new device that isolates single cells and their progeny, MIT researchers can now trace detailed family histories for several generations of cells descended from one “ancestor.”
This technique, which can track changes in gene expression as cells differentiate, could be particularly useful for studying how stem cells or immune cells mature. It could also shed light on how cancer develops.
“Existing methods allow for snapshot measurements of single-cell gene expression, which can provide very in-depth information. What this new approach offers is the ability to track cells over multiple generations and put this information in the context of a cell’s lineal history,” says Robert Kimmerling, a graduate student in biological engineering and the lead author of a paper describing the technique in the Jan. 6 issue of Nature Communications.
The paper’s senior authors are Scott Manalis, the Andrew and Erna Viterbi Professor of Biological Engineering and a member of MIT’s Koch Institute for Integrative Cancer Research, and Alex Shalek, the Hermann L.F. von Helmholtz Career Development Assistant Professor of Health Sciences and Technology, an assistant professor of chemistry, and a member of MIT’s Institute for Medical Engineering and Science.
Capturing cell lineage
The new method incorporates a recently developed technology called single-cell RNA-seq, which sequences the messenger RNA from a single cell. These RNAs, known collectively as the transcriptome, reveal which genes are being actively transcribed (that is, copied into messenger RNA instructions for building proteins) inside a cell at a given point in time. This helps scientists understand, for example, what makes a skin cell so different from a heart cell even though the cells share the same DNA.
“Scientists have well established methods for resolving diverse subsets of a population, but one thing that’s not very well worked out is how this diversity is generated. That’s the key question we were targeting: how a single founding cell gives rise to very diverse progeny,” Kimmerling says.
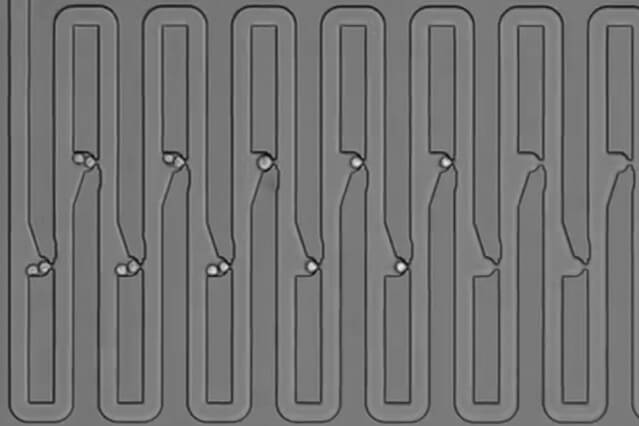
To try to answer that question, the researchers designed a microfluidic device that traps first an individual cell and then all of its descendants. The device has several connected channels, each of which has a trap that can capture a single cell. After the initial cell divides, its daughter cells flow further along the device and get trapped in the next channel. The researchers showed that they can capture up to five generations of cells this way and keep track of their relationships.
To get the cells off the chip, the researchers temporarily reverse the direction of the fluid flowing across the chip, allowing them to remove the cells one at a time to perform single-cell RNA-seq.
In this study, the researchers captured and sequenced immune cells called T cells. These cells are on constant alert in the body, and when they encounter a cell infected with a virus or bacterium, they leap into action, creating two distinct populations — effector T cells, which seek and destroy infected cells, and memory T cells, which retain a memory of the encounter and circulate in the body indefinitely in case of a subsequent encounter.
“A single founding cell can give rise to both effector and memory cell subtypes, but how that diversity is generated isn’t very clear,” Kimmerling says.
The researchers analyzed RNA from recently activated T cells and two subsequent generations. When comparing genes with functions related to T cell activation and differentiation, they found that “sister” cells produced from the same division event are much more similar in their gene expression profiles than two unrelated cells. They also found that “cousin” cells, which have the same “grandmother,” are more similar than unrelated cells, which suggests unique, family-specific transcriptional profiles for single T cells.
The researchers hope that future studies with this device could help to resolve the long-standing debate over how T cells differentiate into effector cells and memory cells. One theory is that the distinction occurs as early as the first T cell division following activation, while a competing theory suggests that the distinction happens later on, as a result of changes in the cells’ microenvironment. To address this question, the researchers believe they would need to analyze the development of T cells taken from a mouse that had been exposed to a foreign pathogen, which would provide a useful model of T cell activation in response to infection.
The new device could also be used to link RNA transcriptome information with other cell traits, the researchers say.
“It opens up possibilities that have never been there before,” Manalis says. “We can further annotate single-cell transcriptome data by applying this strategy to our existing devices for measuring mass, growth rate, density, or deformability.”
“I think this is really beautiful work,” says Dean Felsher, a professor of medicine and pathology at Stanford University School of Medicine. “It builds on what Scott has been doing for a while, which is creating a whole new way of interrogating single-cell measurements. Now he can follow the progeny over multiple generations, which is really hard to do.”
Cellular “age”
In this study, the researchers also discovered that they could use their new technique to learn which genes are expressed at certain points during the cell division cycle. Because they trap each cell and have a record of when it last divided, they can directly link the “age” of each cell to its transcriptome.
They identified a set of about 300 genes that correspond most with time since division (a proxy for cell cycle progression), and found that most of those genes were involved in cell division. Therefore, by measuring the levels of those 300 genes in similar cells, scientists should be able to estimate the ages of those cells. The researchers also found that a leukemia cell line, which proliferates continuously, has a different set of genes that appear to be driving cell division.
“In the future, this approach may be able to provide insight into unique transcriptional regulators of cell cycle progression in various cancer models,” Kimmerling says.