The neurodegenerative disease known as frontotemporal dementia (FTD) causes untold suffering. As neurons die in regions of the brain important for maintaining our personalities and living a purposeful life, patients experience steadily worsening cognitive and behavioral symptoms, and the disease is generally fatal within a decade of diagnosis.
Though FTD is not as well known as Alzheimer’s disease, it’s the second most common cause of dementia in people under 65, and there’s currently no treatment.
For years researchers have been trying to solve the puzzle of just how FTD starts breaking down the brain. They know what the brain looks like after years of damage, but knowing what happens first would help direct the development of drugs to someday treat the disease. Now, in a Nature paper published Aug. 31, 2020, UC San Francisco researchers weigh in on that long-standing key question, laying out a clear timeline of disease in an FTD mouse model and showing that the brain’s immune system is responsible for kicking off the destruction.
In 2016, Eric Huang, MD, PhD, a UCSF professor of pathology, and his team linked FTD to excessive activity in microglia, brain immune cells that normally work to clear debris, fight infection, and help shape neural connections during development. Researchers knew that both microglia and neurons fare poorly in FTD, both eventually marred by the pathological clumping of a protein called TDP-43 – a classic sign of FTD both in mouse models and in many human cases. But whether neurons or microglia are affected first remained a mystery.
Now Huang’s team has finally answered that question, examining a disease-relevant part of the brain called the thalamus and looking cell-by-cell to see how gene expression changes as mice age and FTD ravages the brain. The work showed how a common human FTD mutation could drive TDP-43 accumulation and the death of vulnerable neurons in the thalamus, with diseased microglia serving as executioners.
“Before neurons show any abnormalities, microglia are already showing signs of disease,” said Huang, who’s affiliated with the UCSF Weill Institute for Neurosciences and is a pathologist at the UCSF-affiliated San Francisco VA Medical Center. “We’ve nailed down a very important mechanism relevant to a devastating human disease.”
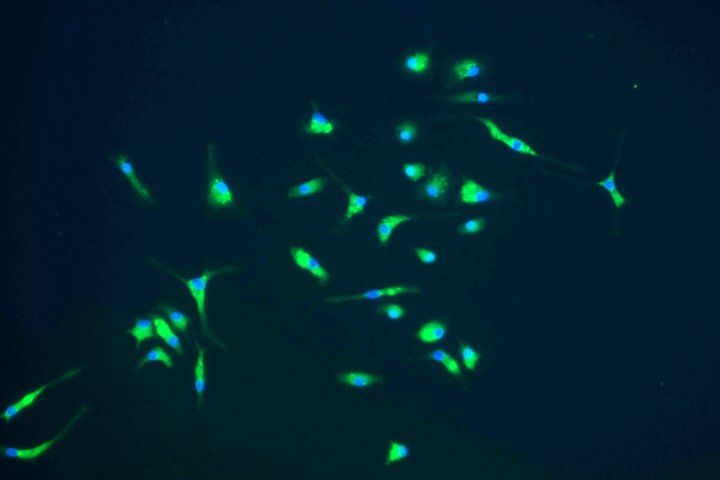
The team also demonstrated precisely how diseased microglia inflict their damage in thalamic neurons, which are important for relaying information through the brain. To study this, the team grew neurons in a fluid that had previously contained microglia from mice with FTD caused by a lack of the progranulin gene (GRN), which is mutated in about 15 percent of familial FTD cases in people. They discovered that exposure to this fluid caused TDP-43 to accumulate and neurons to begin to die, suggesting that the microglia must have secreted something into the fluid that was toxic to the neurons.
The researchers narrowed the culprits down to immune molecules called C1qa and C3, which they also showed could drive TDP-43 build-up and kill neurons. The team confirmed this by showing that exposure to progranulin-deficient microglia that had been engineered not to produce these specific molecules no longer posed a threat to neural health.
With the path of destruction clearly laid out by the new findings, Huang and his team are now working to determine what kind of drugs might restore health to progranulin-deficient microglia and thus potentially protect the brains of FTD patients. They also hope to examine how signs of microglial disease could serve as biomarkers showing whether candidate FTD drugs are actually working.
Many approaches to FTD so far have focused on how treatments affect neurons, but Huang says that’s looking too late in the game – those drugs would treat symptoms and not the cause of the disease.
“It’s time to focus on drugs for microglia,” he said.
