Hematology researchers at The Children’s Hospital of Philadelphia have manipulated key biological events in adult blood cells to produce a form of hemoglobin normally absent after the newborn period. Because this fetal hemoglobin is unaffected by the genetic defect in sickle cell disease (SCD), the cell culture findings may open the door to a new therapy for the debilitating blood disorder.
“Our study shows the power of a technique called forced chromatin looping in reprogramming gene expression in blood-forming cells,” said hematology researcher Jeremy W. Rupon, M.D., Ph.D., of The Children’s Hospital of Philadelphia. “If we can translate this approach to humans, we may enable new treatment options for patients.”
Rupon presented the team’s findings today at a press conference during the annual meeting of the American Society of Hematology (ASH) in New Orleans. Rupon worked in collaboration with a former postdoctoral fellow, Wulan Deng, Ph.D., in the laboratory of Gerd Blobel, M.D., Ph.D.
Hematologists have long sought to reactivate fetal hemoglobin as a treatment for children and adults with SCD, the painful, sometimes life-threatening genetic disorder that deforms red blood cells and disrupts normal circulation.
In the normal course of development, a biological switch flips during the production of hemoglobin, the oxygen-carrying component of red blood cells. Regulatory elements in DNA shift the body from producing the fetal form of hemoglobin to producing the adult form instead. This transition occurs shortly after birth. When patients with SCD undergo this transition, their inherited gene mutation distorts adult hemoglobin, forcing red blood cells to assume a sickled shape.
In the current study, Rupon and Blobel reprogrammed gene expression to reverse the biological switch, causing cells to resume producing fetal hemoglobin, which is not affected by the SCD mutation, and produces normally shaped red blood cells.
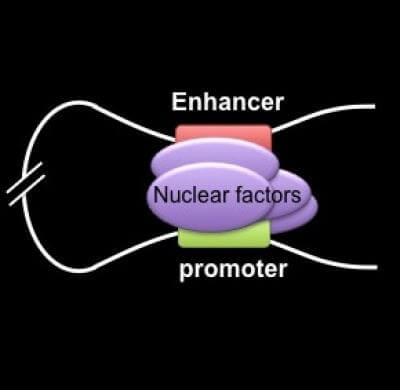
The scientists built on previous work by Blobel’s team showing that chromatin looping, a tightly regulated interaction between widely separated DNA sequences, drives gene transcription—the conversion of DNA code into RNA messages to carry out biological processes.
In the current study, the researchers used a specialized tool, a genetically engineered zinc finger (ZF) protein, which they custom-designed to latch onto a specific DNA site carrying the code for fetal hemoglobin. They attached the ZF to another protein that forced a chromatin loop to form. The loop then activated gene expression that produced embryonic hemoglobin in blood-forming cells from adult mice. The team obtained similar results in human adult red blood cells, forcing the cells to produce fetal hemoglobin.
Rupon and Blobel will continue investigations aimed at moving their research toward clinical application. Rupon added that the approach may also prove useful in treating other diseases of hemoglobin, such as thalassemia.
