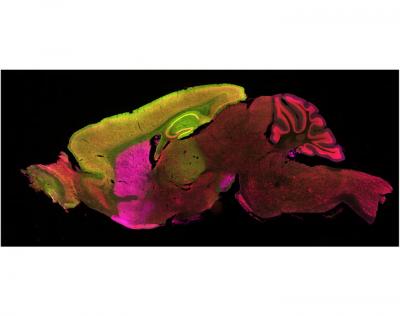
The gene mutation that causes Huntington’s disease appears in every cell in the body, yet kills only two types of brain cells. Why? UCLA scientists used a unique approach to switch the gene off in individual brain regions and zero in on those that play a role in causing the disease in mice.
Published in the April 28 online edition of Nature Medicine, the research sheds light on where Huntington’s starts in the brain. It also suggests new targets and routes for therapeutic drugs to slow the devastating disease, which strikes an estimated 35,000 Americans.
“From day one of conception, the mutant gene that causes Huntington’s appears everywhere in the body, including every cell in the brain,” explained X. William Yang, professor of psychiatry and biobehavioral sciences at the Semel Institute for Neuroscience and Human Behavior at UCLA. “Before we can develop effective strategies to treat the disorder, we need to first identify where it starts and how it ravages the brain.”
Huntington’s disease is passed from parent to child through a mutation in a gene called huntingtin. Scientists blame a genetic “stutter” — a repetitive stretch of DNA at one end of the altered gene—for the cell death and brain atrophy that progressively deprives patients of their ability to move, speak, eat and think clearly. No cure exists, and people with aggressive cases may die in as little as 10 years.
Huntington’s disease targets cells in two brain regions for destruction: the cortex and the striatum. Far more neurons die in the striatum—a cerebral region named after its striped layers of gray and white matter. But it’s unclear whether cortical neurons play a role in the disease, including striatal neurons’ malfunction and death.
Yang’s team used a unique approach to uncover where the mutant gene wreaks the most damage in the brain.
In 2008, Yang collaborated with co-first author Michelle Gray, a former UCLA postdoctoral researcher now at the University of Alabama, to engineer a mouse model for Huntington’s disease. The scientists inserted the entire human huntintin gene, including the stutter, into the mouse genome. As the animals’ brains atrophied, the mice developed motor and psychiatric-like problems similar to the human patients.
In the current study, Yang and Nan Wang, co-first author and UCLA postdoctoral researcher, took the model one step further. They integrated a “genetic scissors” that snipped off the stutter and shut down the defective gene—first in the cortical neurons, then the striatal neurons and finally in both sets of cells. In each case, they measured how the mutant gene influenced disease development in the cells and affected the animals’ brain atrophy, motor and psychiatric-like symptoms.
“The genetic scissors gave us the power to study the role of any cell type in Huntington’s,” said Wang. “We were surprised to learn that cortical neurons play a key role in initiating aspects of the disease in the brain.”
The UCLA team discovered that reducing huntingtin in the cortex partially improved the animals’ symptoms. More importantly, shutting down mutant huntingtin in both the cortical and striatal neurons–while leaving it untouched in the rest of the brain– corrected every symptom they measured in the mice, including motor and psychiatric-like behavioral impairment and brain atrophy.
“We have evidence that the gene mutation highjacks communication between the cortical and striatal neurons,” explained Yang. “Reducing the defective gene in the cortex normalized this communication and helped lessen the disease’s impact on the striatum.”
“Our research helps to shed lights on an age-old question in the field,” he added. “Where does Huntington’s disease start? Equally important, our findings provide crucial insights on where to target therapies to reduce mutant gene levels in the brain–we should target both cortical and striatal neurons.”
Some of the current experimental therapies can be delivered only to limited brain areas, because their properties do not allow them to broadly spread in the brain.
The UCLA team’s next step will be to study how mutant huntingtin affects cortical and striatal neurons’ function and communication, and to identify therapeutic targets that may normalize cellular miscommunication to help slow progression of the disease.
Why don’t they just use some kind of immunotherapy to remove mutant huntingtin?
I hope nanotechnology technology will be advanced enough to treat such disorders in the near future. It is very sad how easily it can be fatal to aggressive people. It is not time for geneticists to work extra hard on coming up with ways to prevent such traits to be transfared from parents to their offspring.
Genetic Scissors is truly the scientific tool of the future in medical manipulation and scientific research. Genetic Scissors are also known as ‘Zinc-finger nucleases’ and consist of restriction enzymes. These enzymes can cut out a piece of a genome which can then be deleted or moved to other organisms such as mice in order study more successfully. It can also be used to add new fragments into the genome. This method of gene scissors has successfully been used to remove the gene coding for insulin which is then placed in bacteria. The bacteria then subsequently produce insulin. Apart from all the diseases mentioned by other comments this can also be used to more successfully research into AIDS. The UCLA team deserves praise!
It is truly incredible, since scientists are already so close to curing this horrific disease. A single injection of an “antisense” drug slowed and partially reversed progression of the disease in affected mice and monkeys.
This drug is effective as it interferes with and blocks out the protein-making “instructions” from the Huntington’s gene.
This is extremely good news!
I also think that further study of the Human Genome Project could result in the cure of Huntington’s disease and other diseases, such as Alzheimer’s disease and Cystic Fibrosis, by scientists learning how to deactivate some of the infected genes.
What makes Huntingtons Disease so dreadful is its neurodegenerative nature. Sufferers experience a deteriorating quality of life owing to their decline in cognition and muscle coordination and they also experience psychiatric episodes.
The current approach in treating Huntington’s Disease is to treat the symptoms and not the disease itself.
However owing to Yang and her colleagues’ inventive approach we are able to make new discoveries. The development of the ‘genetic scissors’ enables scientists to enhance their understanding of the afflicted gene. This improved understanding will initiate investigators to embark on new research with an aim towards developing a cure and not merely treating the symptoms.This research will also accelerate the rate at which a cure can be found.
The exciting aspect of science is that it is not limited. Scientists can merge their research to create pioneering ideas. Fore example genetic engineering bio-technology can be combined to develop devices that will directly slow progression, improve the cells ability to survive harmful effects and replace damaged neurons. This can be applied to all sorts of genetic diseases such as Cystic Fibrosis and Alzheimers to name a few. There are no limits to science and its valuable contributions to our lives!
This could turn into a very interesting story. I would also like to find out more about the “genetic scissors”. If this could be a new way of treating genetic disorders, the UCLA team should really try to take that next step so that we can be more informed about the situation. Maybe they can also find a way to cure this disorder before it is past on to children. I think the method of using mice to find out more of the Huntington’s disease is very clever because mice are similar to humans.
Mice are one of the best animals to use for these experiments. Not only because their genes are similar to humans but also because their easy to observe and contain, and the results of these experiments take affect quite quickly.
It is impeccable to learn that such amazing breakthroughs are still taking place in the scientific world today. I hope to be a part of it someday.
The development of genetic scissors is a very effective way of reducing huntington ‘s disease. Such developments of new ways for destroying or rather reducing certain diseases in the human body is a clear indication that there are new developments and strategies that are created in the medical world to combat all sorts of disease.
The use of mice is a very important feature when trying to combat a disease in a human body since most of the genes that are found in mice are almost similar to those of human beings.
The research being done by the UCLA team is truly ground breaking. Most of the experiments were done on mice, however it would be interesting to know id the effects are the same in humans. I am also interested to know if this treatment can help with the effect of motor neuron disease of which there is very little known.
That fact that the disease is passed past parents to offspring, that it is a genetic disease..makes it interesting. I also think that the UCLA team are taking the right path of finding a way to reduce the Huntington’s disease by first learning it’s role and origin in the brain.
This article was very interesting. I feel that this technique, such as the use of the genetic scissors, is the beginning of a whole new way of treating genetic disorders and types of dementia in particular. It is a very innovative technique! Huntington’s disease is a devastating disease that causes so much anguish and pain in many people’s lives. Since it is genetic, it must cause so much worry in the children of these patients to know that there is a possibility of having the gene themselves, especially after watching their parents suffer through it. Therefore I think it is amazing that because of this research, scientists may be able to prolong the symptoms and affects. I wonder if they could even use the same technique with Alzheimer’s disease, an equally devastating type of Dementia? I am also wondering what the genetic scissors consist of. Perhaps some form of restriction enzymes?
I am curious about the “genetic scissors”.What do they consist of and would this method of treatment be successful in relieving or curing other neuron associated diseases for example amyotrophic lateral sclerosis (ALS)?