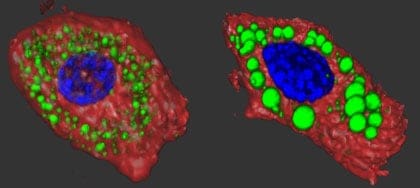
Practically everyone gets fatter as they get older, but some people can blame their genes for the extra padding. Researchers have shown that two different mutations in a gene called ankyrin-B cause cells to suck up glucose faster than normal, fattening them up and eventually triggering the type of diabetes linked to obesity.
The more severe of the two mutations, called R1788W, is carried by nearly one million Americans. The milder mutation, known as L1622I, is shared by seven percent of the African American population and is about as common as the trait for sickle cell anemia.
The findings, which were generated in mice, could help identify at-risk individuals who might be able to tip the scales back in their favor by eating better and exercising more. The results appear July 13 in the Journal of Clinical Investigation.
“This is one of the first examples of a susceptibility gene that would only be manifested through a modern lifestyle,” said Vann Bennett, M.D., Ph.D., senior author of the study and George Barth Geller Professor of Biochemistry, Cell Biology, and Neurobiology at Duke University School of Medicine. “The obesity epidemic really took off in the 1980’s, when sugary sodas and French fries became popular. It’s not like we suddenly changed genetically in 1980, but rather we have carried susceptibility genes that were exacerbated by this new diet. We think our findings are just the beginning, and that there are going to be many genes like this.”
Bennett, who is also an investigator with the Howard Hughes Medical Institute, discovered ankyrin-B more than thirty years ago. He found that ankyrin-B acts as a kind of protein anchor, tethering important proteins to the inside of the cell’s plasma membrane. Since his initial discovery, Bennett and other researchers have implicated defects in ankyrin-B in a wide variety of human afflictions, including irregular heartbeat, autism, muscular dystrophy, aging, and, more recently, diabetes.
Diabetes is quickly becoming one of the greatest threats to public health, as waistlines expand around the world and here in the United States. If the current trends continue, one in three Americans will have diabetes by 2050. Patients with type 1 diabetes do not make enough insulin, the hormone that helps process the glucose that builds up in the bloodstream after a meal. Patients with type 2 diabetes, the form linked to obesity, make insulin but become resistant to its effects.
Several years ago, the Bennett laboratory found evidence that ankyrin-B mutations might play a role in insulin secretion and metabolism. Since then, several studies have uncovered rare ankyrin-B variants that are associated with type 2 diabetes. One mutation, called R1788W, was more common in Caucasians and Hispanics. Another, called L1622I, was found exclusively in African-Americans, a group known to be at a particularly high risk of diabetes. But it was still unclear how these changes in the genetic code could set a course for diabetes.
To get at that answer, Bennett’s MD/PhD student Jane Healy created mouse models that carried these same human genetic variants. She and her colleagues found that animals with two copies of the R1788W mutation made less insulin than normal mice. Despite this shortcoming, their blood glucose levels were normal. So the researchers performed the rodent equivalent of a glucose tolerance test –- commonly used to screen for type 2 diabetes in people — to determine how quickly glucose was cleared from the bloodstream in the mutant mice. To their surprise, the mutant mice metabolized glucose more quickly than normal mice.
“We thought that the main problem in these mice would be with the beta cells that produced and secreted insulin,” said Healy, co-author of the study and a former trainee in Bennett’s laboratory. “Instead, our most significant finding lay with the target cells, which took up much more glucose than expected.”
Glucose doesn’t enter cells and tissues all on its own, but instead has to rely on a second molecule, called GLUT4 transporter, to gain access. Normally, GLUT4 hangs out in the cell, like a hostess waiting for party guests to arrive. When insulin is present it acts as a kind of doorbell, alerting GLUT4 to spring into action and open the door to let glucose into the cell. When insulin goes away, the GLUT4 transporters close the door, turn around, and go back into the middle of the cell.
However, postdoctoral fellow Damaris Lorenzo, Ph.D., found that wasn’t the case with the mutant mice. After conducting a number of biochemistry experiments, Lorenzo ddiscovered that the mice had lots of GLUT4 on the surface of their muscle and fat cells even when there wasn’t any insulin around. That meant that glucose could flow in without necessarily having to bother with the doorbell.
This open door policy was an advantage when they were young, because it protected the animals from low insulin levels. But when the mice got older — or switched to a particularly high-fat diet — it made the mice fatter and, eventually, led them to become insulin resistant.
The researchers believe that long ago, the R1788W mutation — and the milder L1622I mutation — may have provided an evolutionary advantage. Aging hunter-gatherer types, who weren’t as effective at chasing down their next meal, needed to gain as much fat as possible to avoid starvation. Now that high-fat, high-calorie foods are plentiful in much of the world, these variants put people at increased risk for modern afflictions like obesity and diabetes.
“If people with these mutations are detected early enough, they become prime candidates for intervention with personalized therapies.” said Lorenzo, lead author of the study. “That might involve specific strategies to manage their deficits in insulin secretion, as well as adhering to a normal diet and an active lifestyle, with the hope that they can avoid the metabolic diseases that could severely impair their quality of life.”
Next, the researchers would like to explore whether the effects they observed in mice hold true in humans. They plan to genotype people in the general population, identify families with ankyrin mutations, and then perform family histories as well as glucose metabolism tests to assess the consequences of these genetic variants at a cellular level.
The research was supported by the Howard Hughes Medical Institute and the George Barth Geller Professorship fund.