A University of Michigan Health System laboratory study reveals a key trigger for producing normal red blood cells that could lead to a new treatment for sickle cell disease.
The study, conducted in mice, appears in this week’s early edition of the Proceedings of the National Academy of Sciences, and holds promise for preventing the painful episodes and organ damage that are common complications of sickle cell disease.
According to the U-M study, increasing the expression of the proteins, TR2 and TR4, more than doubled the level of fetal hemoglobin produced in sickle cell mice and reduced organ damage.
“The vast majority of sickle cell disease patients are diagnosed early in childhood when adult hemoglobin normally replaces fetal hemoglobin, but the severity of the disease can differ markedly, correlating most strongly with the level of fetal hemoglobin present in red cells,” says pediatrician and lead study author Andrew D. Campbell, M.D., director of the Pediatric Comprehensive Sickle Cell Program at the U-M Comprehensive Cancer Center.
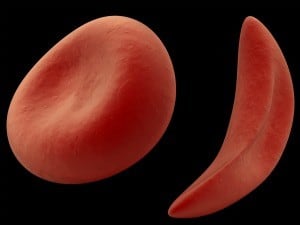
Sickle cell is an inherited blood disorder impacting millions of patients worldwide that causes normal red blood cells to change shape to a crescent moon.
The result is debilitating pain episodes, chronic organ damage and a significantly shortened life span.
Sickle cell disease occurs in 1 out of every 500 African American births and 1 out of every 36,000 Hispanic American births, according to the Sickle Cell Disease Association of America, Inc.
It’s most common among those with an ancestry to Africa, South and Central America, the Caribbean islands, India, Saudi Arabia and Mediterranean countries such as Turkey, Greece and Italy.
But a small number of sickle cell patients are born with a high enough fetal hemoglobin level to moderate complications.
The study team, that included experts in hematology oncology, cell and developmental biology and pathology from the U-M and the University of Tsukuba, Japan, demonstrated a potential method for boosting the fetal hemoglobin levels by modulating TR2/TR4 expression.
“While the average fetal hemoglobin was 7.6 percent in the sickle cell mice, the TR2/TR4 treated sickle cell mice had an average fetal hemoglobin of 18.6 percent,” says senior study author James Douglas Engel, Ph.D. , professor and chair of the U-M’s Cell and Development Biology Department.
It’s the first time specific proteins have been targeted to prevent a disease, he says.
Anemia and red blood cell turnover all improved within the TR2/TR4 treated mice. Additional studies, including clinical trials, would be required to determine if the method could help humans.
“Currently hydroxyurea is the only FDA approved drug known to increase the levels of fetal hemoglobin within sickle cell disease patients and a substantial number of patients do respond to it,” says Campbell, the pediatric hematology oncology specialist. “But the long term consequences for hydroxyurea are unknown, especially in children.”
###
Authors: Andrew D. Campbell, Shuaiying Cui, Lihong Shi, Rebekah Urbonya, Andrea Mathias, Kori Bradley, Kwaku O. Bonsua, Rhonda R. Douglas, Brittne Halford, Lindsay Schmidt, David Harro, Donald Giacherio, Keiji Tanimoto, Osamu Tanabe, and James Douglas Engel.
Reference: “Forced TR2/TR4 Expression in Sickle Cell Disease Mice Confers Enhanced Fetal Hemoglobin Synthesis and Alleviated Disease Phenotypes,” Proceedings of the National Academy of Sciences, Oct. 31, 2011.
Funding: Work by authors is supported by the American Heart Association, Cooley’s Anemia Foundation, Robert Wood Johnson Foundation and the National Institutes of Health’s National Heart Lung and Blood Institute.
Resources
University of Michigan Department of Cell and Developmental Biology
http://www.med.umich.edu/cdb/
U-M Sickle Cell Program
http://www.med.umich.edu/sicklecell/
U-M Comprehensive Cancer Center
http://www.cancer.med.umich.edu/