Disrupting a problematic gene in brain cells can reverse Huntington’s disease pathology and motor symptoms in a mouse model of the inherited neurological disorder, Emory scientists report.
The researchers used CRISPR/Cas9 gene editing, delivered by a viral vector, to snip part of a gene producing toxic protein aggregates in the brains of 9-month old mice. Weeks later, where the vector was applied, aggregated proteins had almost disappeared. In addition, the motor abilities of the mice had improved, although not to the level of control mice.
The results were published June 19, 2017 in Journal of Clinical Investigation.
The findings open up an avenue for treating Huntington’s as well as other inherited neurodegenerative diseases, although more testing of safety and long-term effects is needed, says senior author Xiao-Jiang Li, MD, PhD, distinguished professor of human genetics at Emory University School of Medicine.
Huntington’s disease is caused by a gene encoding a toxic protein (mutant huntingtin or mHTT) that causes brain cells to die. Symptoms commonly appear in mid-life and include uncontrolled movements, balance problems, mood swings and cognitive decline.
Touted widely for its potential, CRISPR/Cas9 gene editing has not been used to treat any neurodegenerative disease in humans. Several concerns need to be addressed before its use, such as effective delivery and the safety of tinkering with DNA in brain cells. A similar approach, but using a different technology (zinc finger nucleases), was reported for Huntington’s disease in 2012.
The mice used in this study have a human mutant huntingtin gene replacing one of the mouse huntingtin genes. In these mice, motor problems and aggregated mutant huntingtin can be observed around the age of 9 months.
When planning gene editing, the scientists selected guide sequences that targeted both the normal copy and the disease-driving copy of the huntingtin gene. This “non-allele specific” approach would not need to be customized to the patient’s genome, unlike other gene editing proposals for Huntington’s disease.
The Emory researchers have previously shown that mice older than four months do not need the huntingtin gene to stay healthy, suggesting that treatment strategies that aim to shut off both copies of the gene in adult humans could be safe. Clinical studies have begun of such treatments, which probably will require continuous administration of the gene-silencing drug. In contrast, a gene editing treatment could be more durable, if it hits enough cells.
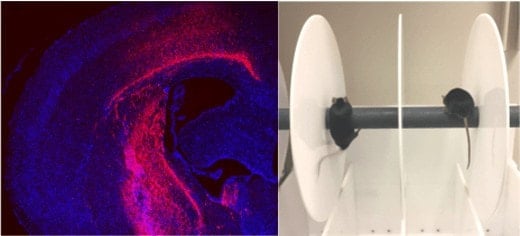
To get CRISPR/Cas9-guided enzymes into brain cells, the researchers harnessed a widely used gene therapy vehicle based on AAV (adeno-associated virus). The scientists injected viral vectors carrying CRISPR/Cas9 into the striatum region of the brains of Huntington’s disease model mice at the age of 9 months. The striatum is a region of the brain that controls body movement and motor function.
This led to a “dramatic decrease” in aggregated mutant huntingtin in the striatum three weeks later. The study reveals the capacity of brain cells to heal themselves if the genetic source of the toxic proteins is removed, the scientists say.
In comparison with control Huntington’s mice, CRISPR/Cas9-injected mice showed significant improvements on tests of motor control, balance and grip strength, although they did not recover to the point where they performed as well as control mice.
Addressing genetic safety concerns, the researchers showed that in brain cells, frameshift mutations triggered by CRISPR/Cas9 occurred predominantly within the huntingtin gene and not in other potential off-target genes. However, the long-term effects and safety of injecting AAV in the brain to express CRISPR/Cas9 remain to be rigorously tested before applying this approach to patients, Li says.
The co-first authors of the paper are postdoctoral fellows Su Yang, PhD at Emory University and Renbao Chang, PhD at Institute of Genetics and Developmental Biology, Chinese Academy of Sciences.
Emory co-authors include Zhaohui Qin, PhD, associate professor of biostatistics, Peng Jin, PhD, professor of human genetics, and Shihua Li. MD, professor of human genetics. Xiao-Jiang Li also is affiliated with the Guangdong-Hongkong-Macau Institute of CNS Regeneration, Jinan University.
The research was supported by the National Institute of Neurological Disorders and Stroke (NS036232, NS101701, NS095279) and the National Natural Science Foundation of China (grant 91332206).
If our reporting has informed or inspired you, please consider making a donation. Every contribution, no matter the size, empowers us to continue delivering accurate, engaging, and trustworthy science and medical news. Independent journalism requires time, effort, and resources—your support ensures we can keep uncovering the stories that matter most to you.
Join us in making knowledge accessible and impactful. Thank you for standing with us!