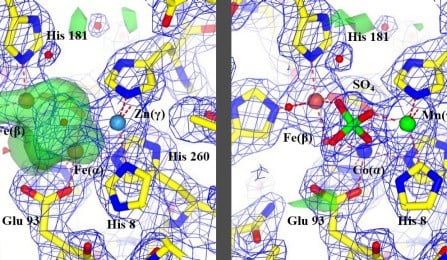
Proteins that contain metal, known as metalloproteins, play important roles in biology, regulating various pathways in the body, which often become targets for lifesaving drugs. While the amount of metal in such proteins is usually tiny, it is crucial to determining the function of these complex molecules.
Scientists have long known that metalloproteins are vital to understanding diseases, such as cancer, and for developing new drugs since inhibitors of metalloproteins have been used to treat diseases from cancer and HIV/AIDS to bacterial infections and hypertension. But there hasn’t been a reliable, analytical method for determining the identity and quantity of metal atoms in metalloproteins.
Now, in a study published last month in the Journal of the American Chemical Society, an international team of researchers report that they have developed a way to unambiguously identify and count metal atoms in proteins in an efficient and routine way. Using it, the team — which included scientists from the University at Buffalo, Hauptman-Woodward Medical Research Institute and others— revealed new information that was there, but previously hidden.
Called Particle Induced Emission of X-rays, or PIXE, the method was first developed in the 1990s by Elspeth F. Garman of the University of Oxford and Geoffrey W. Grime of the University of Surrey Ion Beam Centre, both authors on the current paper.
The breakthrough reported in the current paper is the development of the method into an efficient high-throughput approach and the combination with other experimental data to identify the type and accurate position of the metals in the proteins. This allows many different types of proteins, large numbers of which make up life as we know it, to be analyzed quickly and efficiently, and provides new information for a better structural understanding.
The team applied the new method to 30 randomly selected metalloproteins, which are already in the global repository of protein structures called the Protein Data Bank. What happened next shocked them.
‘The result was stunning’
“I sat in Buffalo with my collaborator from Oxford and when we crunched the numbers, we both immediately realized we’d made a discovery,” recalled Edward Snell, PhD, one of the corresponding authors, who is president and CEO of Hauptman-Woodward and professor in the Department of Materials Design and Innovation, a joint program of UB’s School of Engineering and Applied Sciences and its College of Arts and Sciences. “We turned the numbers into a picture and hidden within the data was an explanation of how this molecular machine worked.
“We were the first in the world to see what had been hiding there all the time. The result was stunning.”
The results showed that the methods previously used to determine some of these 30 random protein structures had either misidentified the metal atom or, in some cases, completely missed it.
“According to our results, the current knowledge of about half of the samples we studied is incorrect,” said Snell.
The researchers noted that the Protein Data Bank is a critical resource for researchers worldwide. In 2017, there were on average 1.86 million downloads per day in the U.S. alone. They note that an enormous number of researchers use structures from the data bank without knowledge of the potential fundamental errors that may be present.
And currently, more than 30% of the data bank models contain a metal.
Profound implications
“Extrapolating from our results in which there was a misidentified metal in at least half of the samples studied suggests that over 350,000 models downloaded per day may not contain the correct metal,” Snell said. “This has profound implications for those using the models. If these models are wrong, the understanding of the millions of people who use them becomes flawed.”
Snell explained that one of the difficulties in studying metals in proteins is that they are very sensitive to X-ray radiation, so the experiment itself can change what you see. But he noted, a technique using X-Ray Free Electron Lasers (XFELs), prevents this because the experiments are usually faster than any change that can occur.
Snell directs the National Science Foundation BioXFEL Science and Technology Center, (Biology with X-ray Free Electron Lasers) a consortium of UB, Hauptman-Woodward and their partners. The center is dedicated to using XFELs, which produce incredibly intense X-rays in extremely short pulses, and can help in the accurate understanding of these metals in biological systems.
Based on his experience with Hauptman-Woodward’s high-throughput crystallization screening center, Snell collaborated to implement the PIXE technique in a high-throughput setting. He used his knowledge of X-ray properties to identify that new structural information was present in the data and then took that knowledge and turned it into a structural result.
“Basically, my colleagues identified the metals and our work in Buffalo showed them where to put them, revealing the new information that became available when the metal in the model was correct,” he said.
In addition to Snell, Garman and Grime, other co-authors on the paper are Oliver B. Zeldin and Edward D. Lowe of Oxford University; Mary E. Snell of Hauptman-Woodward; John F. Hunt and Liang Tong of Columbia University; and Gaetano T. Montelione of Rensselaer Polytechnic Institute.
The research in the U.S. was funded by the National Science Foundation BioXFEL Science and Technology Center.
Media Contact Information
Ellen Goldbaum
News Content Manager
Medicine
Tel: 716-645-4605
[email protected]
Twitter: @UBmednews
