Brown fat is a tissue that runs hot. Packed with mitochondria, threaded through with blood vessels, laced with nerve fibers that fire when the temperature drops, it is, in metabolic terms, a furnace, and researchers have spent the better part of two decades trying to work out how to light it. The ambition is straightforward enough: a tissue that burns glucose and lipids to generate heat, rather than storing them, looks like a promising target for obesity treatments that work on energy expenditure rather than appetite suppression. What has been harder to pin down is the infrastructure problem, the question of what actually keeps brown fat wired up and running. A new study published in Nature Communications suggests the answer starts with a single protein that splits in two.
Farnaz Shamsi’s lab at NYU College of Dentistry has been working on the internal communication networks of brown adipose tissue for several years, and what they’ve found changes the picture considerably. It’s not enough, it turns out, to simply have brown fat. The tissue’s heat-producing capacity depends critically on maintaining dense networks of blood vessels and sympathetic nerves, and those networks need to be actively built and maintained by the fat cells themselves.
Brown fat functions through a feedback loop that is, in some respects, rather elegant. When the brain senses cold, it signals through the sympathetic nervous system, releasing norepinephrine into brown adipose tissue. The norepinephrine triggers thermogenesis, the process of generating heat from stored fuels, and the blood vessels supply oxygen and substrates to keep it going. But chronic cold exposure also drives the tissue to expand: more brown adipocytes, more capillaries, more nerve branches, all growing in coordination to sustain the response. The question, for a long time, was what coordinates that expansion. Shamsi’s team had previously identified a signaling molecule called SLIT3 as a likely candidate; the new paper establishes exactly how it works, and why it can do two jobs at once.
SLIT proteins are not new to biology. They were first characterized as axon guidance molecules, cues that help developing neurons find their way to the right destinations during brain development. The finding that one of them operates deep inside fat tissue, organizing blood vessel growth and nerve branching, is somewhat unexpected. In a way, it suggests the body has repurposed an old piece of molecular machinery for a rather different task.
The key to SLIT3’s dual function is that it doesn’t stay whole. An enzyme called BMP1, expressed in the fat cell precursors that secrete SLIT3, cleaves the protein into two structurally distinct fragments: a larger N-terminal piece and a smaller C-terminal piece. This cleavage turns out to matter enormously. The two fragments bind to different receptors on different cell types and drive different processes: one promotes the growth of new blood vessels, while the other recruits and expands sympathetic nerve fibers. “It works as a split signal, which is an elegant evolutionary design in which two components of a single factor independently regulate distinct processes that must be tightly coordinated in space and time,” said Shamsi. Neither fragment, acting alone, can replicate what the intact system achieves.
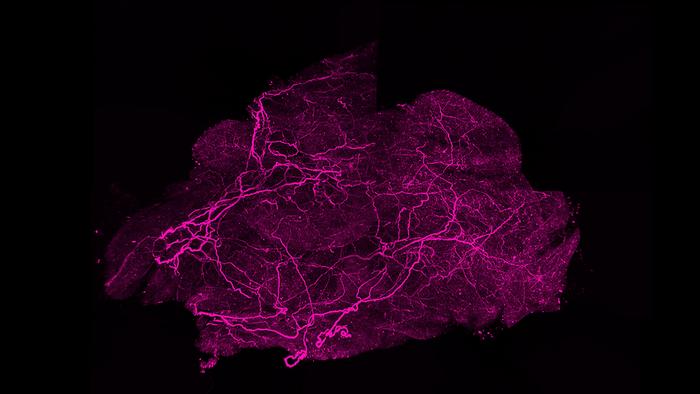
The researchers tracked this through a series of experiments in mice, using a gene-silencing approach to knock out SLIT3 specifically in brown fat tissue. The results were striking. Mice lacking SLIT3 in their brown fat struggled badly in the cold, unable to maintain normal body temperature. Their brown fat, examined after a week of cold exposure, had undergone a kind of metabolic collapse: the tissue whitened, accumulated lipid droplets, and showed dramatically reduced expression of UCP1, the protein most directly responsible for heat generation. Three-dimensional imaging of the nerve architecture, using a tissue-clearing technique that renders the tissue transparent and allows lightsheet microscopy to map the entire volume, showed that the sympathetic nerve network had become sparse and disorganized.
Adults do have brown fat, though considerably less of it than infants, and its activity varies substantially between individuals. Studies using PET scanning have found active brown fat in the neck and upper chest of lean, cold-exposed adults, and people with more active brown fat tend to burn more calories at rest. Whether it’s possible to meaningfully increase brown fat activity through drugs or lifestyle changes, enough to produce clinically significant weight loss, remains an open question that researchers are actively working on.
The appetite pathway has historically been easier to intervene on. GLP-1 receptor agonists, for instance, work through well-characterized receptors in the gut and brain to reduce hunger and slow gastric emptying, a mechanism that was understood well enough to develop into drugs relatively quickly. Energy expenditure through tissues like brown fat involves more complex signaling, requires precise targeting, and the field has lacked the detailed mechanistic understanding to design effective interventions. The new SLIT3 findings are partly valuable because they provide that kind of mechanistic detail.
This is essentially the question the NYU study addresses. Brown fat function depends not just on the fat cells themselves but on the network of blood vessels that supply oxygen and fuel, and the sympathetic nerve fibers that tell the tissue when to switch on. Without adequate innervation, the fat cells don’t receive the norepinephrine signal that triggers thermogenesis; without enough blood vessels, they can’t sustain the metabolic activity even if they’re activated. Research in people with obesity has found evidence of disrupted sympathetic innervation in fat tissue, which may partly explain why thermogenic function is impaired even when the cells appear structurally intact.
The short answer is that it doesn’t, exactly. SLIT3 gets cleaved into two structurally different fragments by an enzyme called BMP1, and each fragment then acts independently on a different cell type through a different receptor. The larger fragment promotes blood vessel growth; the smaller fragment drives nerve branching. Both are produced from the same original protein, by the same fat progenitor cells, at the same time, which is what allows the two processes to be coordinated in space and timing without requiring two separate signals.
What the experiments also showed is that the fat cells themselves, at least their intrinsic thermogenic machinery, remained intact. When the researchers bypassed the nervous system entirely, administering a compound that directly activates adrenergic receptors in the tissue, the brown fat responded normally, cranking up its thermogenic gene programs as if SLIT3 had never been removed. The defect, in other words, is not inside the fat cells. It’s in the wiring. This distinction matters for how to think about brown fat therapeutics, and perhaps for why some people’s brown fat functions poorly even when the cells themselves appear structurally normal.
The C-terminal fragment, SLIT3-C, turns out to signal through a receptor called PLXNA1, which sits on the surface of sympathetic nerve fibers in brown fat. PLXNA1 is a plexin, a type of receptor previously known mainly for responding to semaphorin proteins during axon development, so its role here extends it into new territory. Knocking down PLXNA1 produced essentially the same sparse innervation phenotype as removing SLIT3 entirely, and mice that overexpress SLIT3-C in their brown fat, but lack PLXNA1, cannot use that extra SLIT3-C to boost their heat production. The N-terminal fragment, meanwhile, works through a different receptor, ROBO4, expressed exclusively on blood vessel cells, driving capillary growth by a separate pathway. Two fragments, two receptors, two processes, all originating from one precursor protein secreted by fat cell progenitors.
Whether this translates to human biology is always the question, and here the team had access to something useful: tissue samples from roughly 1,500 people enrolled in the Leipzig Obesity BioBank, a large German cohort with detailed metabolic characterizations. SLIT3 gene expression in fat tissue correlated with markers of metabolic health, including circulating adiponectin levels and macrophage content in visceral fat depots. “That really got our attention, as it suggests that this pathway could be relevant in human obesity and metabolic health,” said Shamsi. Genome-wide association studies had already flagged variants near the SLIT3 gene as associated with insulin resistance and higher BMI; the correlation data gives that genetic signal a possible mechanism.
The study also identifies BMP1 as the first vertebrate SLIT protease, a finding with implications beyond fat tissue. SLIT proteins regulate cell migration, angiogenesis, and inflammatory responses across many tissue types, and understanding how they’re processed into fragments raises questions about whether the same splitting logic applies elsewhere, in tumor biology, nerve regeneration, wound healing. That the fragment identities were unknown while full-length SLIT proteins had been studied for decades is a reminder of how many molecular details remain obscure even in well-characterized systems.
For obesity research specifically, the findings push back against a simple view of brown fat activation. Stimulating fat cells to produce heat is one thing; maintaining the infrastructure that allows them to receive signals, access fuel, and distribute warmth throughout the body is quite another. The nerve and vessel networks aren’t passive bystanders that happen to be present. They’re actively constructed, through molecular crosstalk between fat progenitor cells and the non-fat cells in the tissue, and without that construction the furnace can’t run at full capacity regardless of how healthy the individual adipocytes might be. Current weight loss drugs, including the GLP-1 receptor agonists that have dominated recent clinical attention, work primarily by reducing food intake. A therapy that works on the expenditure side, through brown fat, would operate by a different mechanism entirely.
Whether SLIT3, BMP1, or PLXNA1 could be targeted therapeutically remains to be established. The biology is complex enough that simple interventions are probably some distance away. But the study maps a pathway that was previously invisible, and in doing so, it redefines what “activating brown fat” might eventually mean.
DOI / Source: https://doi.org/10.1038/s41467-026-70310-9
ScienceBlog.com has no paywalls, no sponsored content, and no agenda beyond getting the science right. Every story here is written to inform, not to impress an advertiser or push a point of view.
Good science journalism takes time — reading the papers, checking the claims, finding researchers who can put findings in context. We do that work because we think it matters.
If you find this site useful, consider supporting it with a donation. Even a few dollars a month helps keep the coverage independent and free for everyone.