Key Takeaways
- Researchers discovered a rare genetic condition linked to a mutation in the IVNS1ABP gene, resulting in cognitive and motor skill decline in affected children.
- They used advanced techniques like exome sequencing and cellular reprogramming to understand the mutation’s impact on neural development.
- The mutation disrupts actin dynamics during cell division, causing cells to enter a zombie-like state called cellular senescence.
- Applying chemicals to stabilize actin structure showed promise in improving cell division rates, suggesting a potential treatment avenue.
- This approach highlights the value of combining genomic analysis with cellular models to explore and treat rare genetic diseases.
The first clue was the hair. A family had come to the attention of Bruno Reversade’s genetics team in Singapore, and among the teenagers were children whose hair had begun to turn white. That alone might have been unremarkable, a quirk, a cosmetic oddity. But these same children were losing motor skills, struggling cognitively, falling behind in ways that had nothing to do with ordinary adolescent difficulty. Reversade’s team reached out to Su-Chun Zhang, a neuroscientist at Sanford Burnham Prebys in California. What they were seeing, Zhang suspected, was something medicine had simply never encountered before.
Progeria syndromes, the cluster of rare genetic conditions that cause children to age decades ahead of schedule, have a well-known feature: they tend to spare the brain. Children with Hutchinson-Gilford progeria, probably the most studied of these conditions, typically retain their cognitive faculties even as their bodies age catastrophically. The neurological and intellectual deficits in Reversade’s family of patients didn’t fit. “Cognitive functions are often well-preserved in these conditions,” Zhang said, “so it was clear from the patients’ progressive loss of motor skills and neurological and intellectual deficits that this was an unknown disease.”
The team turned to genomics. Using exome sequencing combined with a mapping technique for recessive traits, they traced the disease to a mutation in a gene called IVNS1ABP. The name is a mouthful, and what it encodes is, on the face of it, baffling: a protein that normally helps influenza viruses hijack a cell’s RNA splicing machinery. The protein had been studied in the context of flu. That was more or less the whole of its scientific biography. Nobody had ever linked it to aging, or to nerve cells, or to anything resembling what this family was experiencing.
Fang Yuan, a staff scientist in Zhang’s lab and first author on the paper, published this month in Nature Communications, put it plainly. “Relatively little research has been done on this gene and protein, and no one has ever linked them to the biology of aging, premature aging diseases or neuropathy,” she said. “It was a mystery in many ways, and one we were determined to solve.”
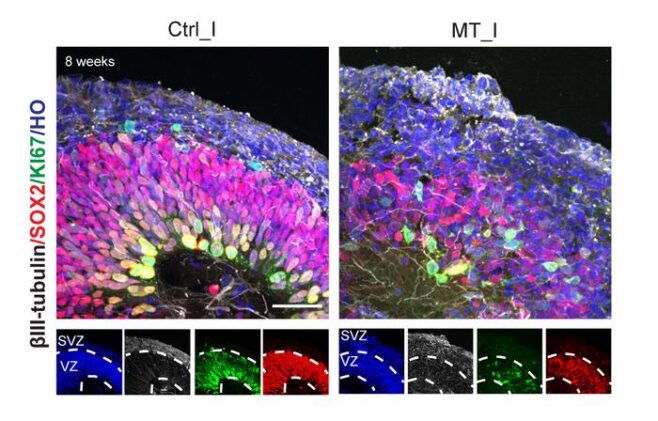
What the team did next is worth dwelling on, because the method is almost as significant as the finding. They took skin samples from affected patients, reprogrammed those cells into induced pluripotent stem cells (iPSCs), and then coaxed those stem cells partway toward becoming neural progenitor cells, the intermediate stage between a stem cell and a functioning neuron. These cells still carry the mutation, which meant the researchers could essentially run experiments on the patients’ disease in a dish. The comparison group was reprogrammed from a sibling without the mutation. It is a technique that has transformed rare disease research over the past decade, though it remains demanding to execute well.
Under the microscope, the difference was immediate. “We found that the patient-derived cells with the mutation grow much slower compared to the control group reprogrammed from a sibling without the disease,” Zhang said. Slow-growing cells are a canonical sign of cellular senescence, the zombie-like state in which a cell stops dividing but refuses to die, churning out inflammatory signals that damage surrounding tissue. Three separate markers of DNA damage were elevated in the patient cells, along with raised expression of CDKN2A, a gene that acts as a kind of brake on the cell cycle. The cells were, in a sense, burning out before they’d really started.
That’s the puzzle at the heart of this discovery. IVNS1ABP was originally identified because it interacts with influenza virus proteins, but its core biological role turns out to be in regulating actin, the structural protein that scaffolds cells. When the IVNS1ABP gene is mutated, actin assembly during cell division goes wrong in ways that have nothing to do with flu and everything to do with how cells age and die. The flu connection, it seems, was a red herring in the gene’s naming history.
They enter a state called cellular senescence, sometimes described as a zombie mode: the cell stops replicating but doesn’t die, and instead leaks inflammatory signals that damage surrounding tissue. In the IVNS1ABP patients, this appears to happen in neural progenitor cells, the precursors that are supposed to expand and mature into neurons during brain development. When those cells senesce prematurely, the brain’s architecture can be disrupted before it has properly formed.
The researchers have a cautious early signal. Applying chemicals that stabilize actin structure in patient-derived cells improved the rate of normal cell division, suggesting the mechanism is at least potentially correctable. That result is in a cellular model, not a patient, and the team is still developing an animal model to test whether it holds. The distance from a cell-culture finding to an approved therapy is long, but having a clear molecular target at this early stage is more than most newly discovered rare diseases can claim.
They used two complementary genomic approaches: exome sequencing to read the protein-coding regions of the patients’ DNA, and a technique for mapping recessive traits that helped narrow the search. From there, they used skin cells from affected patients, reprogrammed them into stem cells, and differentiated those stem cells into neural progenitor cells that still carry the mutation. That gave them a living cellular model of the disease to experiment on, rather than having to infer everything from DNA sequence alone.
Possibly, though with caveats. Cellular senescence and disrupted actin dynamics are both implicated in normal aging biology, not just in rare genetic conditions. Whether the IVNS1ABP pathway plays any role in the gradual accumulation of senescent cells that characterizes typical aging is an open question the researchers haven’t yet addressed. Rare progeroid syndromes have historically offered disproportionate insight into how aging works in general, so it wouldn’t be the first time a vanishingly rare disease pointed toward something universal.
Tracing why took some patience. Because IVNS1ABP had no known direct connection to cell division, the researchers suspected the mutation was doing its damage indirectly, through a chain of protein interactions they hadn’t yet mapped. A proteomics screen turned up 14 candidate proteins that might be involved. Ten of them were associated with actin, the structural protein that gives cells their shape. That pointed toward cytokinesis, the final physical act of cell division, when a cell must pinch itself in two. During this process, actin filaments have to assemble into a contractile ring, a tight, even, circular structure that draws inward to sever the cell. Zhang described the geometry with some precision: it normally forms a very round and even ring. In patient cells, that ring was shrunken and irregular, a warped hoop instead of a clean circle, meaning cells were being pulled apart asymmetrically and sustaining damage in the process. “When these actin dynamics are altered,” Yuan said, “the cell cannot perform cell division at the right time and in the right place.”
The consequences ripple outward in ways you might expect and some you might not. Dividing cells that repeatedly botch the final split accumulate chromosomal damage, which triggers senescence. In neural progenitor cells, the effect may be particularly damaging, since the developing brain depends on a carefully choreographed expansion of precursors into the right kinds of neurons at the right stages. The team’s brain organoids, three-dimensional clusters of tissue derived from patient cells, showed a more disorganized architecture and fewer properly patterned developing neurons than healthy controls, consistent with impaired neurodevelopment rather than just accelerated aging.
There is, cautiously, a treatment signal. When the researchers applied chemicals that stabilize actin structure, the rate of normal cell division improved. It’s a cellular-model result, not a therapeutic claim, and Zhang’s team is still developing an animal model to test whether the intervention holds up in vivo. But it suggests the mechanism is, at least in principle, tractable. “We already showed that if we correct some of the steps in the molecular processes, then we can fix some of the defects, at least in the cellular model,” Yuan said.
The broader significance of the work is perhaps methodological as much as medical. The disease itself is vanishingly rare; this appears to be the first family documented with this particular mutation. But the approach, combining genome sequencing with cellular reprogramming to define an unknown disease and then probe potential treatments in the same model system, is applicable to the long tail of rare genetic conditions that remain poorly understood precisely because they’re too rare to study conventionally. Yuan put it in perhaps the most useful framing: “It will be important to complement these findings with studies in an animal model we’re developing, but what we’ve done already demonstrates that this approach is a powerful tool for defining new diseases and developing potential treatments.”
Somewhere in the genetics literature, there are probably other families, other unexplained presentations of premature aging that don’t fit established syndromes, other children whose hair is doing things it shouldn’t. Whether the IVNS1ABP pathway turns out to be a narrow curiosity or something with wider relevance to aging biology, even in its typical form, is a question that researchers are only beginning to ask.
DOI / Source: https://doi.org/10.1038/s41467-026-70756-x
ScienceBlog.com has no paywalls, no sponsored content, and no agenda beyond getting the science right. Every story here is written to inform, not to impress an advertiser or push a point of view.
Good science journalism takes time — reading the papers, checking the claims, finding researchers who can put findings in context. We do that work because we think it matters.
If you find this site useful, consider supporting it with a donation. Even a few dollars a month helps keep the coverage independent and free for everyone.