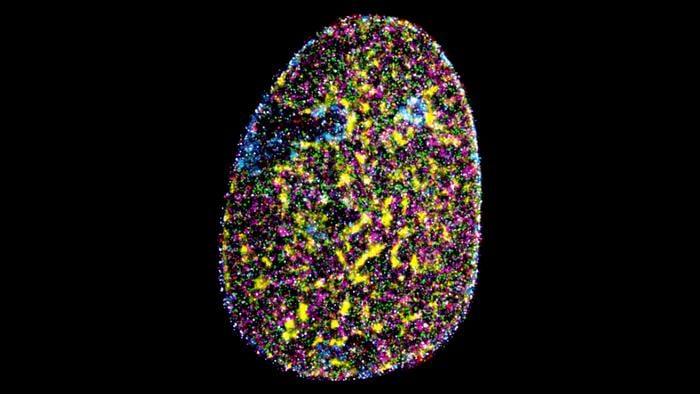
Nine proteins. One nucleus. Four hours.
That’s the thumbnail version of what a team at the Indian Institute of Science in Bangalore has pulled off, and it perhaps undersells the achievement a little. Until recently, the best super-resolution microscopes could track only two or three molecular species at once inside a living cell; what the IISc team has done, essentially, is turn a spotlight into a switchboard. Their modified version of a technique called DNA-PAINT can now illuminate nine different nuclear targets in a single imaging session, mapping their precise locations to within 3 to 5 nanometres, and it does so quickly enough to be practically useful. The results, published last week in Nature Communications, offer something close to a first proper street map of the cell nucleus.
The human cell nucleus is densely crowded in ways that no one has yet fully reckoned with. Trillions of cells, each crammed with millions of proteins, nucleic acids, and molecular machines, all operating in three dimensions, in real time, at scales that defeat conventional microscopes. Mahipal Ganji, who leads the lab at IISc’s Department of Biochemistry, argues that building new ways to see inside individual cells is essential to push the boundaries of biological research. The problem is that seeing inside that crowd has always required compromise: stain for one protein, lose another.
Blinking Beacons and Slow Chemistry
DNA-PAINT works by attaching tiny fluorescent DNA fragments to specific targets inside the cell. Shine a laser through, and each labelled molecule blinks on and off like a miniature beacon, its blinking pattern distinct enough to reconstruct a sharp image of where it sits. The clever part is that different proteins can be tagged with different DNA sequences, so the microscope can, in principle, read multiple signals at once, or in rapid succession. In practice, though, the approach has been hampered by slow chemistry: standard DNA tags bind so sluggishly to their targets that imaging a single protein could take hours. “Conventional techniques are limited to only seeing three molecular species,” says Micky Anand, a PhD student at IISc and co-first author of the study. The bottleneck is not the optics. It’s the molecules.
Ganji’s team set about redesigning those molecules from scratch.
The key was expanding the pool of speed-optimised DNA tags at the heart of the technique. They systematically explored combinations of DNA building blocks, looking for pairs that bind faster and hold on longer, without cross-reacting with each other. Starting from six previously known fast-binding sequences, they identified six more, giving them twelve to work with. Five of the new sequences showed notably longer binding times than the older ones, which turns out to matter for image quality: a tag that lingers on its target for 678 milliseconds produces a sharper localisation signal than one that detaches after 282 milliseconds, partly because the longer-held event generates more photons per frame. The upshot is that some targets can now be imaged at lower laser intensities, reducing the photodamage that has long plagued live-cell experiments.
Abhinav Banerjee, a postdoctoral researcher now at the Janelia Research Campus in Virginia and co-first author, notes that shifting from a few target molecules to nine could open pathways to detecting illness before symptoms emerge. He argues that mapping protein locations at this scale could open up new ways to detect illness before symptoms appear. The logic is roughly this: disease often begins not as a single broken protein but as a pattern, a spatial relationship gone wrong, a cluster that should be dispersed or a gap that should be filled. You can’t see patterns if you can only look at one thing at a time.
Nine Targets, One Cell
To demonstrate the approach in fixed cells, the team labelled nine distinct nuclear marks in HeLa cancer cells, the well-characterised lab strain derived from a cervical tumour, and imaged the lot in under four hours. The nine targets spanned both the active regions of the nucleus, where genes are being read out into RNA, and the quiet heterochromatin zones packed against the nuclear membrane. They included the molecular machinery of transcription, structural proteins forming the nuclear lamina, histone modifications that tag DNA as either accessible or closed off, and SC35, a marker for nuclear speckles, which are the small, poorly understood organelles where RNA splicing happens. Seeing all of these at once, in the same cell, at nanometre resolution, is something no previous imaging system has managed.
The map they produced confirmed several expected relationships. Actively transcribing genes clustered near nuclear speckles. Compacted, silent chromatin hugged the nuclear lamina. But the technique also let the team disturb the system and watch what happened. When they treated cells with Actinomycin D, a drug that blocks RNA synthesis, the nuclear architecture shifted in ways that would have been invisible to conventional microscopes: the amorphous, irregular shape of nuclear speckles collapsed into tighter spheres, and the contacts between active chromatin marks and speckles were lost across the board.
What the Map Does Not Yet Show
There are limits. The technique still requires fixed cells rather than living ones, meaning it captures snapshots rather than movies. And it scales to perhaps ten or twelve simultaneous targets before orthogonality problems, essentially different DNA tags accidentally cross-reacting, start to compromise the data. Eighteen might be achievable, the authors suggest, by combining their approach with a newer variant that uses left-handed DNA sequences.
Still, ten is considerably more than three. The value of simultaneous multiprotein mapping, as opposed to imaging one target after another across different cells, is that it preserves the spatial relationships as they actually exist in a single nucleus, not averaged across populations. No two cells are identical, and disease, in particular, often shows up first in individual outliers. Mapping one nucleus comprehensively, in a single session, at nanometre precision, gets you much closer to the biology that actually matters.
For now, the nine-target map of the HeLa nucleus is something like a first city plan for a territory that was previously only sketched in outline. More proteins will be added; the technique will presumably be adapted for other cell types and, eventually, for primary human samples rather than cell lines. What comes into view when you can see everything at once, rather than one thing at a time, remains an open question.
DOI: 10.1038/s41467-026-72206-0
Frequently Asked Questions
Why does it matter that scientists can see nine proteins at once rather than two or three?
Biology inside a cell is fundamentally about relationships, not individual parts. Seeing only two or three proteins at a time is a bit like trying to understand a city’s traffic patterns by watching one intersection. Nine targets simultaneously, in the same nucleus, preserves the spatial context that gets lost when you image different proteins across different cells and then try to stitch the picture together. That context is especially important for disease, where the telling change is often a pattern rather than a single broken molecule.
How does DNA-PAINT actually work?
The technique works by attaching short, fluorescent DNA fragments to specific proteins inside the cell. Under a laser, these fragments bind and unbind rapidly, creating a distinctive blinking signal that software can use to pinpoint each molecule’s location far more precisely than conventional light microscopy allows. By using different DNA sequences for different target proteins, and swapping imaging solutions between rounds, researchers can build up a multi-layered map of the nucleus without the targets interfering with each other.
Could this approach eventually help diagnose cancer earlier?
That’s the underlying hope. If researchers can establish what a normal nuclear protein map looks like, they can start identifying the subtle spatial disruptions that appear in diseased cells, potentially before any other symptom is detectable. The current technique works on fixed cells rather than live biopsies, so there are several engineering steps between here and a clinical tool, but the spatial mapping principle is sound and the precision is already approaching what diagnosis would require.
What stops the technique from imaging even more proteins simultaneously?
The main constraint is orthogonality: each DNA tag sequence must not accidentally bind to any other target’s complementary strand. The researchers found that two of their twelve new sequences did cross-react slightly, effectively limiting the reliable multiplexing to ten targets per experiment. Extending to eighteen may be possible using left-handed DNA, which has a mirror-image chemical structure and therefore an entirely separate set of non-cross-reacting sequences, but that work is still ahead.
Why were HeLa cells used for this experiment?
HeLa cells are among the most thoroughly characterised cell lines in biology, derived from a cervical tumour in 1951 and cultured continuously ever since. That familiarity is the point: because so much is already known about how their nuclear proteins are supposed to be arranged, the team could verify that their nine-target maps were producing accurate results rather than imaging artefacts. The technique will need to be tested in primary human cells and disease models before its full diagnostic potential becomes clear.
ScienceBlog.com has no paywalls, no sponsored content, and no agenda beyond getting the science right. Every story here is written to inform, not to impress an advertiser or push a point of view.
Good science journalism takes time — reading the papers, checking the claims, finding researchers who can put findings in context. We do that work because we think it matters.
If you find this site useful, consider supporting it with a donation. Even a few dollars a month helps keep the coverage independent and free for everyone.