The pancreas sends its message in a wave of insulin, released into a short stretch of blood vessel called the portal vein that runs directly to the liver. Under normal conditions the liver soaks up roughly 80 percent of that signal before it reaches anywhere else in the body, calibrating blood sugar with a precision that most of us never think about. But researchers at the National Institutes of Health have now discovered that hepatitis C virus does something peculiar to this communication channel: it suppresses the message at source, apparently coaxing the pancreas to secrete less insulin in the first place, while also muffling the liver’s ability to extract what little does arrive. The downstream result, paradoxically, is that blood sugar and insulin levels in the general circulation look completely normal.
That last detail is what makes the finding striking. People with chronic hepatitis C infection have long been known to develop insulin resistance and, in some cases, type 2 diabetes, but the standard blood tests would not have flagged anything obviously wrong in the patients studied here.
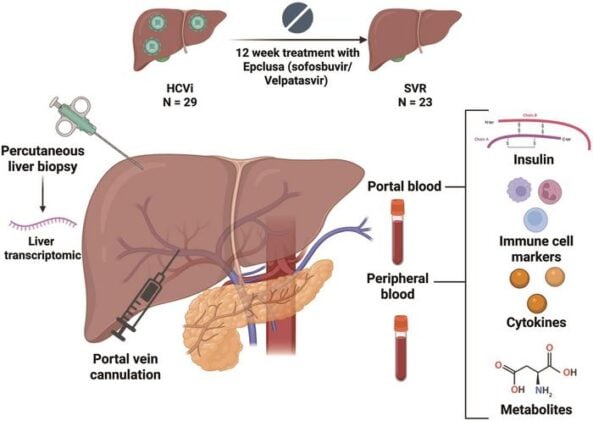
The team, led by Theo Heller at the NIH Clinical Center, recruited 29 people with chronic HCV infection and followed them through a course of direct-acting antiviral treatment, sofosbuvir and velpatasvir, re-evaluating 23 of them a year after they had achieved a sustained virologic response, meaning the virus was gone. What made the study unusual was where the researchers took their blood samples. As well as the standard peripheral draw from the arm, they collected blood directly from the portal vein, giving them a view of insulin dynamics that previous studies had simply not attempted. They also took liver biopsies and performed comprehensive immunological, metabolomic, and transcriptomic analyses at both time points. It is, as studies of this kind go, a fairly invasive protocol, which is perhaps why no one had done it quite like this before.
Portal insulin was roughly half the level during active infection compared to after viral clearance. And yet peripheral insulin, peripheral glucose, portal glucose: none of it shifted significantly. This is the puzzle.
The explanation the researchers favour involves two simultaneous failures. First, the virus seems to reduce how much insulin the pancreas secretes into the portal vein. Second, the liver, already inflamed and transcriptionally reprogrammed by the infection, extracts less of what arrives, leaving peripheral concentrations artificially propped up. The two effects roughly cancel out in the blood tests that most clinicians would actually order, which could mean that metabolic disruption is quietly accumulating in HCV patients who look, on paper, metabolically fine. Standard measures of insulin resistance, including the commonly used HOMA-IR score, showed no significant change across the infection period in this cohort.
The immune system’s fingerprints were all over the portal insulin signal. Among 65 serum markers tested, proinflammatory cytokines including TNF-alpha and IL-17 correlated positively with portal insulin during infection but not after the virus was cleared. Vascular injury markers told a similar story. The percentage of naive cytotoxic T-cells in both portal and peripheral blood moved in the opposite direction from portal insulin, suggesting that as the immune response activated this particular lymphocyte population, the insulin supply to the liver fell. Natural killer cells, by contrast, tracked positively with portal insulin both during and after infection, a relationship that seems to persist independently of HCV status and may reflect something more fundamental about how the innate immune system interacts with insulin signalling.
Liver transcriptomics added another layer. Of 71 gene pathways that correlated significantly with portal insulin during infection, 20 out of 21 immune pathways correlated positively, while metabolic and amino acid pathways mostly correlated negatively. Key genes in the insulin resistance pathway, JNK1 and IKKB, were upregulated during infection. The O-GlcNAc regulatory pathway, which promotes insulin resistance in high-glucose environments, was also activated. None of this showed up in blood glucose measurements, which stayed stable throughout, but the liver’s molecular machinery was apparently behaving as though insulin resistance conditions were in play regardless.
Three unusual nucleotides complicated things further (in the way that unexpected metabolomics findings tend to complicate things). Orotate, a pyrimidine precursor that has separately been shown to help maintain pancreatic beta-cell function in diabetic mouse models, was lower in portal blood during infection. Two modified nucleosides, t6A and 7-methylguanine, both correlated with portal insulin during infection. These are molecules involved in translational regulation and RNA modification, and the researchers speculate that reduced insulin signalling may be disrupting hepatic RNA turnover in ways that could ripple outward. The honest caveat here is that the causal direction is unclear; these are correlations from a small, albeit carefully collected, dataset.
The study’s limitations are real and the authors are straightforward about them. Twenty-nine patients is not many, and the absence of C-peptide measurements, which would have let the team calculate hepatic insulin extraction more precisely, is a gap. Pancreatic biopsies were not taken. The findings are correlational and hypothesis-generating rather than mechanistically proven.
Still, the clinical implications are plausible enough to take seriously. If HCV can suppress portal insulin through immune mechanisms before conventional markers deteriorate, then patients may be accumulating metabolic risk invisibly, including those who are not yet cirrhotic and who might not be prioritised for metabolic monitoring. The researchers suggest that successful antiviral treatment, by clearing the virus and reducing hepatic inflammation, could improve insulin dynamics independently of any weight change, a potentially useful lever in a condition where metabolic complications drive a lot of long-term morbidity.
There is also a broader question lurking here, one that the authors gesture toward at the end of their paper. HCV has been a remarkably productive model for understanding how viruses interact with liver metabolism, partly because effective treatments now give researchers a clean before-and-after window that most infectious disease studies cannot offer. If the gut-liver axis carries real-time immunometabolic signals that viruses can intercept and distort, the same principle might apply in other hepatic inflammatory conditions. The portal vein, it turns out, is carrying more information than standard tests have ever bothered to read.
Frequently Asked Questions
Why would blood tests look normal if hepatitis C is disrupting insulin signalling?
The virus appears to reduce pancreatic insulin secretion and hepatic insulin extraction at the same time, and the two effects roughly cancel each other out in general circulation. Standard blood tests measure peripheral insulin and glucose, not what is happening in the portal vein between the pancreas and the liver, so the disruption can be essentially invisible using conventional markers. This means metabolic damage could be accumulating in patients who appear, on standard workups, to be metabolically healthy.
Could clearing hepatitis C fix the insulin problem?
The study found that portal insulin levels roughly doubled a year after viral clearance, suggesting the disruption is at least partly reversible. More interestingly, the researchers propose that reducing hepatic inflammation through antiviral treatment may improve insulin dynamics independently of weight loss, which is significant because it implies a metabolic benefit of viral eradication beyond just stopping liver damage. Whether this translates into reduced long-term diabetes risk remains to be tested in larger studies.
Is the pancreas actually being infected by hepatitis C?
HCV infection of pancreatic tissue has been reported in earlier research, and this study’s findings are consistent with some kind of HCV-mediated pancreatic dysfunction. The researchers found that immune changes during infection, particularly in specific T-cell populations, correlated with lower portal insulin in ways that suggest immune-driven suppression of beta-cell secretion. The exact mechanism is not yet established, and pancreatic biopsies were not collected in this study, so direct evidence of infection-related pancreatic damage was not available here.
Why does the HCV genotype seem to matter for insulin levels?
Patients infected with genotype 1 HCV had higher portal insulin during infection than those with other genotypes, particularly genotype 3, which is already associated with more aggressive liver disease and steatosis. This suggests the virus’s strain-specific properties may affect how severely it disrupts the pancreas-liver insulin axis, though the numbers in this study were too small to draw firm conclusions. It is a finding that probably warrants its own dedicated investigation.
ScienceBlog.com has no paywalls, no sponsored content, and no agenda beyond getting the science right. Every story here is written to inform, not to impress an advertiser or push a point of view.
Good science journalism takes time — reading the papers, checking the claims, finding researchers who can put findings in context. We do that work because we think it matters.
If you find this site useful, consider supporting it with a donation. Even a few dollars a month helps keep the coverage independent and free for everyone.