Getting a cancer drug to a tumor is only half the problem. Within hours of arrival, many therapeutics begin to drift, diluted by blood flow, expelled by pumps embedded in cancer cell membranes, or simply diffusing into surrounding healthy tissue before they’ve had a chance to work. The tumor, in other words, doesn’t hold onto them. And that failure of retention, more than anything else in drug development, might explain why treatments that look promising in the lab so often disappoint in the clinic. A team at the University of California, San Francisco has spent years trying to fix this, and their latest results suggest a rather elegant solution: a molecular grappling hook that physically embeds itself in the cancer cell membrane and refuses to let go.
The system, described in a paper published this week in ACS Central Science, works by exploiting one of the tumor’s own biochemical quirks to anchor drugs in place rather than releasing them to wander.
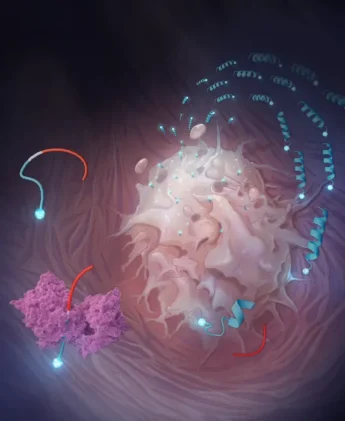
At the heart of the approach is a class of engineered peptides the researchers call restricted interaction peptides, or RIPs. Each one is roughly 30 amino acids long and built in three sections, almost like a Swiss Army knife snapped shut. The first section, derived from an antimicrobial peptide called Temporin L, is the business end: it can fold into a corkscrew-like structure and dig into cell membranes. The second section is a masking domain that keeps the first section folded and inert, prevented by its electrical charge from touching anything. Between them sits the key: a short sequence specifically designed to be cut by a protease enzyme that the tumor produces in abundance. Leave the peptide whole, and the masking domain does its job, keeping the whole assembly inactive. Clip it in the right place, and the membrane-grabbing end springs free.
The protease in question is fibroblast activation protein, or FAP. It’s a serine protease expressed at high levels by cancer-associated fibroblasts, the supportive cells woven through the stroma of solid tumors, present in nearly every cancer type. FAP has long attracted interest as a therapeutic target, partly because it’s so consistently expressed across tumor types and largely absent from normal adult tissue. Early attempts to block it pharmacologically ran into trouble (protease redundancy means blocking one enzyme rarely achieves much), but the new strategy does the opposite: instead of stopping FAP, it uses FAP as a trigger.
Designed to Stick
To build the FAP-activated version, which they call FRIP, the team first needed to know exactly what sequences of amino acids FAP prefers to cut. They screened 228 peptides simultaneously using a technique called multiplex substrate profiling by mass spectrometry, essentially running hundreds of cleavage reactions in parallel and tracking the results by mass. The winner was a sequence reading HIGP-TAAY, which FAP cleaved at a rate roughly comparable to, and in some respects faster than, collagen, FAP’s natural substrate in tissue.
What happened next, when FRIP met a live cancer cell, was visible in real time under a confocal microscope. Fluorescently tagged FRIP added to U-251 glioblastoma cells (a line chosen for its high FAP expression) produced a sharp ring of fluorescence along the cell membrane within about 10 minutes. By 20 minutes, the signal had moved inward: the peptide, and whatever it was carrying, had been swallowed whole. Block FAP with an inhibitor first, and the ring never forms. Use a cell line with low FAP expression, and the effect is sluggish. The mechanism is about as clean as these things get.
For the drug delivery test, the team attached MMAE (monomethyl auristatin E), a potent cell-killing agent widely used in antibody-drug conjugates. The combination proved toxic to glioblastoma cells at concentrations roughly 30 times lower than needed for the undelivered drug, suggesting the membrane-tethering step was shuttling far more payload into cells than passive diffusion alone could manage. Michael Evans, one of the study’s corresponding authors, has argued that retaining drugs within tumors is perhaps the most overlooked dimension of cancer pharmacology. The data rather support him.
Outsmarting the Clinic’s Standard Bearer
In mice bearing head and neck cancer tumors, FRIP loaded with MMAE shrank tumors more effectively than the drug alone, and without the body-weight loss that forced the free-drug group to be withdrawn from the study at day 8. That last detail matters: MMAE is genuinely toxic at the doses needed to reach therapeutic effect systemically. Tethering it to a tumor-specific delivery vehicle appears to substantially reduce the collateral damage, letting the drug concentrate where it’s needed without the rest of the body bearing the consequences.
The team then swapped the chemotherapy payload for radioactive copper, first for imaging (copper-64, a positron emitter) and then for therapy (copper-67, a beta emitter). Here the comparison gets particularly pointed. The researchers pitted their FRIP against FAPI-46, an active site-directed FAP radioligand currently in clinical trials and something of a gold standard in the field. Both compounds cleared from blood at nearly the same rate. But 24 hours after injection, FRIP had accumulated in tumors at roughly five times the concentration of FAPI-46. The underlying reason is almost counterintuitively simple: FAPI-46 binds to FAP in a one-for-one stoichiometric fashion, while FRIP exploits the catalytic nature of the enzyme. A single FAP molecule can clip many FRIP peptides in sequence, each one anchoring to the nearest membrane, building up like barnacles. The competition, by contrast, can only get as far as there are active sites to occupy.
One dose of therapeutic copper-67 loaded onto FRIP delayed glioblastoma growth significantly compared to vehicle and outperformed an equivalent dose of the clinical-stage radioligand. The authors are careful, as they should be, to note that the different chelation chemistry and pharmacokinetics make a completely rigorous comparison difficult. But the signal is hard to ignore.
Evans says the technology should maximize drug delivery to tumor tissue while sparing normal tissues, and that the same molecule used to image a tumor could eventually treat it. First-in-human imaging studies are expected to begin later this year, with a UCSF spinout called Therapaint developing the platform commercially. The version already in clinical trials, GRIP B, targets a different enzyme (granzyme B, released by immune cells in the tumor microenvironment) and is being used to monitor responses to immunotherapy. FRIP is the oncology-targeted version of what is, in principle, an endlessly adaptable system: swap the cleavage sequence, swap the payload, and theoretically you have a new targeting agent for any disease where a protease runs hot.
Whether that modularity holds up in human bodies, where enzymes roam farther from their tumors and individual variation in FAP expression may complicate the picture, remains to be seen. But the logic of the approach, letting the disease itself arm the weapon, is one of those ideas that seems obvious in retrospect. It took quite a lot of biochemistry to make it work.
https://doi.org/10.1021/acscentsci.6c00185
Frequently Asked Questions
Why do cancer drugs often stop working after a few days even when they reach the tumor?
Once a drug reaches a tumor, it still has to stay there long enough to work, and that’s harder than it sounds. Cancer cells express molecular pumps that actively eject small molecules, and blood flow dilutes drugs that diffuse freely through tissue. Most targeted drug delivery systems focus on getting the drug to the right place but don’t address what happens after arrival. The FRIP approach attempts to solve both problems simultaneously, using the tumor’s own enzyme activity to lock the drug in place the moment it arrives.
What makes fibroblast activation protein a good trigger for this system?
FAP is produced at high levels by cancer-associated fibroblasts, a type of supportive cell found in the surrounding tissue of virtually all solid tumors. It’s largely absent from normal adult tissue, which makes it a relatively clean signal for distinguishing tumor from healthy tissue. Unlike targeting a protein expressed directly on cancer cells, which can vary considerably between patients and tumor types, FAP’s expression in the tumor microenvironment is remarkably consistent, making it a more reliable trigger across different cancers.
How does FRIP differ from existing antibody-drug conjugates?
Antibody-drug conjugates also attach cytotoxic payloads to targeting agents, but the antibody circulates for a long time and can cause immune reactions. FRIP is a much smaller peptide, which changes its pharmacology considerably, and it uses a two-step mechanism: the peptide is activated at the tumor site and then physically embeds in cell membranes rather than relying purely on receptor binding. The membrane-tethering step is the key addition, creating a kind of local depot that keeps drug concentration high at the site of action rather than letting it diffuse away.
Could the same platform work for cancers that don’t express FAP?
In principle, yes. The cleavage sequence sitting between the membrane-binding and masking domains is modular and can be swapped out for sequences recognized by other proteases. The researchers have already demonstrated this with granzyme B, an enzyme released by immune cells, which they’ve used to image immune responses to cancer immunotherapy. Any disease state that features elevated activity of a protease with a relatively specific substrate profile is potentially addressable, though each new version would need its own optimization and validation work before clinical use.
What is the significance of FRIP outperforming a clinical-stage radioligand by five times in tumor uptake?
Radioligand therapy works by concentrating radioactive material in tumors, so higher retention translates directly into more radiation delivered to cancer cells and less to surrounding tissue. FAPI-46, the compound FRIP outperformed in the head-to-head comparison, has shown impressive tumor imaging but modest clinical responses in therapy trials, and limited tumor retention is widely suspected as a reason. If FRIP’s mechanism genuinely achieves durable accumulation at five times the concentration, that gap could be the difference between a drug that slows tumor growth briefly and one that produces meaningful, lasting responses.
ScienceBlog.com has no paywalls, no sponsored content, and no agenda beyond getting the science right. Every story here is written to inform, not to impress an advertiser or push a point of view.
Good science journalism takes time — reading the papers, checking the claims, finding researchers who can put findings in context. We do that work because we think it matters.
If you find this site useful, consider supporting it with a donation. Even a few dollars a month helps keep the coverage independent and free for everyone.