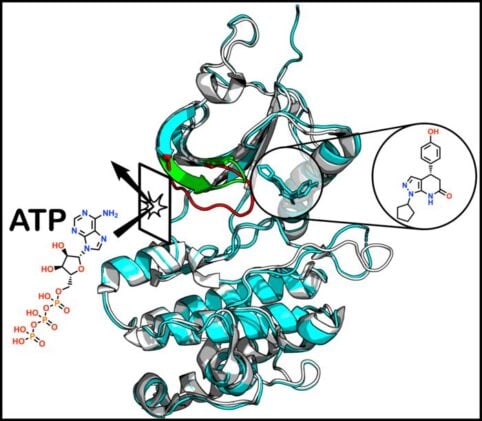
Two molecules sat side by side on the chemists’ screen, near-identical twins. The same scaffold, the same atoms, the same everything, save for a single oxygen-bearing group that had shuffled one position over on a ring. A trivial edit, the sort you might make without thinking. And yet when researchers at the Icahn School of Medicine at Mount Sinai grew crystals of each one locked onto their target protein, the two compounds were doing something completely different.
One had wedged itself into a pocket nobody knew existed. The other behaved itself, sliding into the obvious slot where most drugs of its kind go. That contrast, reported on 2 June in the Journal of the American Chemical Society, is the heart of a study about a cancer protein called PKMYT1, and about how far you can trust artificial intelligence to find the places a drug might bind.
PKMYT1 is a kinase, one of the enzymes that act as molecular switches governing when a cell divides. It sits at a checkpoint late in the cell cycle, holding division back until any DNA damage has been patched up. Healthy cells use this pause sensibly. Tumour cells, it turns out, can lean on it to dodge a self-destruct mechanism that would otherwise kill them off, which is exactly why PKMYT1 has become such an appealing target for new cancer drugs.
The trouble is precision. Nearly every kinase inhibitor on the market jams the same part of the enzyme, the ATP-binding site, where the cell’s energy currency normally docks.
But kinases all share roughly the same ATP pocket, so a drug aimed there tends to hit dozens of the things at once. That’s how you get side effects. The holy grail, for years, has been to find a less crowded, more distinctive spot on the protein, an allosteric site, that only one kinase has.
So the Mount Sinai team went looking with the most powerful tool around. They fed PKMYT1 to AlphaFold2, the AI system that predicts protein shapes, generated a stack of possible conformations, and ran a virtual screen of around 4 million candidate molecules against a pocket the AI suggested might be druggable. A handful of hits came back. One, refined into a compound they call P29, looked worth crystallising.
When the structure came out, it was not what the model had drawn. P29 had bound, yes, but to a site tucked between two structural features called the aC-helix and the P-loop, a cranny that has never been seen occupied in any PKMYT1 structure before. Binding there, P29 nudges a tyrosine residue to swing out into the surrounding solvent and shoves the P-loop into a position that physically blocks ATP from getting in. A previously unobserved way to switch the enzyme off.
The pocket the models missed
“Our study shows both the power and the limitations of AI in drug discovery,” says Avner Schlessinger, who directs the AI Small Molecule Drug Discovery Center at Mount Sinai and co-led the work. “AI was very accurate when predicting known protein shapes, but it missed a completely unexpected binding pocket that we could only uncover experimentally. That hidden site may ultimately provide a new way to design more selective cancer drugs.”
Here is where those identical twins come back in. The team tinkered with P29 to make it stronger, and one tweak, moving a hydroxyl group from the para to the meta position, did the trick: the new compound, P32, was roughly twenty times more potent. It was also, frustratingly, doing the conventional thing, binding the ordinary ATP site rather than the clever hidden one. “One of the most surprising findings was that a very small chemical modification caused the molecule to switch from binding in this hidden pocket to binding in a much more conventional way,” says Michael Lazarus, the study’s other senior author. The takeaway, he adds, is that “these proteins are incredibly dynamic and sensitive to subtle molecular changes. It also reinforces why experimental validation remains essential, even in the era of AI.”
That sensitivity cuts both ways, mind you. P32 may have abandoned the hidden pocket, but it turned out impressively choosy, ignoring all but a few of the forty-odd kinases the researchers tested it against, including the closely related WEE1, and it engaged PKMYT1 inside living cells.
The more sobering result is what happened when the team asked today’s best AI tools to find the hidden site on their own. AlphaFold2, its successor AlphaFold3, and a newer system called Boltz-2 all failed to predict the P29 conformation. Lengthy physics-based simulations stumbled onto something like it only rarely, and at considerable computational expense.
Which doesn’t mean the AI was useless, far from it. The same cofolding tools nailed P32’s conventional binding mode with near-perfect accuracy, and it was an AI model that pointed the team toward the protein’s hidden flexibility in the first place. The models are brilliant at the well-trodden cases and blinkered at the strange ones, presumably because they have learned from a database stuffed with the former and almost none of the latter. A separate pocket-hunting algorithm, run after the fact, could spot the cryptic site lurking in an old structure, which suggests the information was there all along, just not where the headline tools were looking.
For now P29 is a starting point, not a drug; its potency is modest and there’s a long road of optimisation ahead. But the existence of that pocket, and the unusual off-switch it offers, hands medicinal chemists a fresh handle on a protein that badly needs more selective inhibitors. The team now wants to design compounds that commit to the allosteric site rather than drifting back to the ATP pocket, and to go hunting for similar hidden niches in other cancer kinases. If those pockets are as common as this one was unexpected, the lesson may be less about what AI can find and more about what it has, for now, been trained not to see.
DOI / Source: 10.1021/jacs.6c05178
Frequently Asked Questions
Why is it so hard to make a kinase inhibitor that only hits one target?
Most kinase drugs bind the ATP site, the spot where the enzyme grabs the cell’s energy supply, and that site looks almost identical across the hundreds of kinases in the human body. A drug aimed there often blocks many kinases at once, which is a common source of toxicity and side effects. Finding a more distinctive, less conserved pocket unique to one kinase is the long-sought route to cleaner, more selective drugs.
How can two nearly identical molecules bind a protein in completely different ways?
In this study, shifting a single hydroxyl group by one position on a ring was enough to make one compound lock into a newly discovered hidden pocket while its near-twin slotted into the conventional ATP site instead. That happens because the protein itself is constantly flexing between shapes, and tiny chemical changes can tip which shape a molecule prefers. It’s a vivid reminder that proteins are dynamic, not the rigid figures that single snapshots suggest.
Is it true that AI failed to find the new drug pocket?
The leading structure-prediction tools, including AlphaFold2, AlphaFold3 and Boltz-2, did not predict the unusual conformation where the new compound binds, and only experiments revealed it. The same tools were highly accurate on the conventional binding mode, so the failure was specific to the rare, hidden state rather than general. The likeliest explanation is that these models learn from databases dominated by common structures and rarely see exotic ones.
Could this discovery lead to a new cancer treatment?
Potentially, though it’s early days. The compound that exploits the hidden pocket is only modestly potent for now, so it serves as a chemical starting point rather than a finished drug. The real prize is the pocket itself, which gives chemists a fresh, more selective way to switch off a protein implicated in breast, lung, gastric and kidney cancers.
ScienceBlog.com has no paywalls, no sponsored content, and no agenda beyond getting the science right. Every story here is written to inform, not to impress an advertiser or push a point of view.
Good science journalism takes time — reading the papers, checking the claims, finding researchers who can put findings in context. We do that work because we think it matters.
If you find this site useful, consider supporting it with a donation. Even a few dollars a month helps keep the coverage independent and free for everyone.