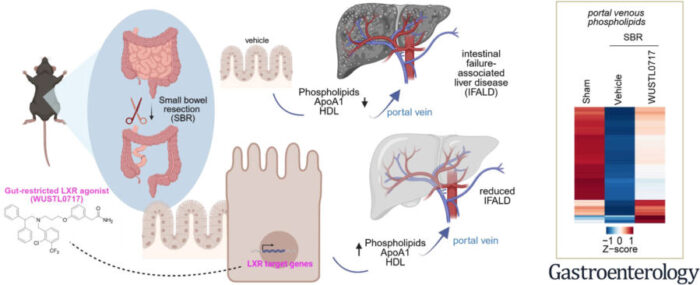
Every day, the small intestine does something extraordinary. Tucked inside its lining, enterocytes (the flat, tile-like cells that line the gut wall) are busy assembling parcels. Each parcel is a tiny sphere of fat and protein, loaded with cholesterol and phospholipids, destined to travel upward through the portal vein into the liver. They’re not just carrying nutrients. They’re carrying a signal: the gut is fine, the liver can relax, everything is under control.
What happens when most of the gut is gone is rather different.
In short bowel syndrome, surgeons remove up to 75% of the small intestine to save a patient’s life, usually following disease that has killed the tissue outright. The surgery works. Patients survive. But those lipid parcels, HDL particles, the “good cholesterol” familiar from cardiovascular medicine, stop arriving in the volumes the liver depends on. Without them, something in the liver shifts. Inflammation builds. Scar tissue accumulates. Over years, the organ that processes everything the remaining gut absorbs begins to fail itself, a condition called intestinal failure-associated liver disease, or IFALD. Up to 15% of patients who’ve undergone major bowel resection develop it, and there is currently nothing, no approved drug, no established protocol, to prevent it once it takes hold.
A new compound synthesised at Washington University in St. Louis could change that.
The drug, called WUSTL0717, was tested in mice that had undergone extensive bowel resection, and the results were striking enough that the research team, led by immunologist Gwendalyn Randolph, is now moving toward larger animal studies. In the treated mice, liver fibrosis was significantly reduced. Weight, almost always lost and rarely recovered after major gut resection, came back at roughly normal rates. And crucially, the drug appears to do all of this while staying where you put it: in the gut, where it’s needed, rather than leaching into the bloodstream and causing the side effects that have sunk every previous attempt at this class of therapy.
“Our goal is to advance a therapeutic drug capable of preserving liver function and mitigating the necessity for liver transplants in people who’ve undergone small bowel surgery,” says Randolph, the Emil R. Unanue Distinguished Professor of Immunology at WashU Medicine. “This study offers a promising pathway for developing such a treatment.”
The history of this story is itself instructive. Liver X receptors, LXRs, a family of proteins inside cells that govern how fat and cholesterol are processed, had long been an attractive drug target for liver disease and atherosclerosis. Activate them, and HDL production goes up. The problem was that LXR agonists, drugs that switch on these receptors, had a habit of switching on too much. They worked in the gut, yes, but they also worked in the liver, triggering a cascade of lipid accumulation that caused rather than cured the liver disease researchers were trying to treat. Several promising compounds failed in clinical trials for exactly this reason.
The solution, in principle, was obvious: find an LXR agonist that stays in the gut and doesn’t reach the liver. In practice, this turned out to be quite hard.
The compound that eventually became WUSTL0717 was first identified by GlaxoSmithKline in 2002, then set aside in favour of a more potent but less selective analogue. Bahaa Elgendy, a medicinal chemist at WashU, synthesised it fresh for this study and ran pharmacokinetic tests to track precisely where it goes after being swallowed. The results were about as good as you could hope for: a strong, short-lived signal in the duodenum, jejunum and ileum; a much weaker signal in the liver; and complete clearance from all tissues within 24 hours. The drug’s chemical structure, an amide group rather than the carboxylic acid group found in its more promiscuous cousin, makes it less soluble, less polar, and apparently less able to escape the intestinal wall into the circulation. It does its job, then the body breaks it down and disposes of it.
“Our future goal is to create the next generation of tissue-specific therapies that preserve therapeutic benefit while reducing unintended systemic effects,” says Elgendy. “This precision-based strategy allows us to revisit important biological targets that were previously considered too challenging to develop safely.”
What the drug actually does in the gut is to turn on genes that produce HDL’s core protein scaffold. ApoA1, the protein backbone of HDL particles, is manufactured by intestinal cells; these cells then load the scaffold with phospholipids particularly phosphatidylcholines and phosphatidylethanolamines and release the assembled particles into the portal blood. When WUSTL0717 activates LXR signalling in the gut lining, ApoA1 production increases, more HDL particles form, and more of them make the journey up the portal vein to the liver. In mice treated with the drug after bowel resection, portal venous levels of both ApoA1 and HDL-cholesterol were higher than in untreated animals. Both measures were inversely correlated with the amount of collagen (the substance of scar tissue) found in the liver.
The team also engineered mice that lack ApoA1 specifically in intestinal cells. These animals fared worse after bowel resection than normal mice: lower portal HDL, more liver damage, more fibrosis. The livers of untreated short bowel mice were accumulating significant amounts of collagen, visible on standard histology stains. In WUSTL0717-treated animals, that collagen accumulation was reversed. Genes associated with the process of liver scarring, including those for collagen synthesis, stellate cell activation, and inflammatory signalling, were measurably quieter.
Perhaps as important as what the drug does is what it doesn’t. Blood triglycerides didn’t rise. Liver fat didn’t accumulate. The drug didn’t deplete neutrophils, a side effect seen with earlier systemic LXR agonists in both mice and humans. Liver enzymes indicating damage remained normal. The intestinal selectivity, in other words, appears to be real and to matter.
None of this has been tested in humans yet. The mouse model of short bowel syndrome, while well-established and informative, involves a version of bowel resection considerably more controlled than the clinical reality. Many patients with short bowel syndrome also receive parenteral nutrition, nutrients delivered directly into the bloodstream, which itself damages the liver, and the research team’s next study will examine whether WUSTL0717 can protect the liver under that additional stress.
The research was partly built on work done by Brad Warner, a paediatric surgeon and researcher at WashU whose career was devoted to improving outcomes for children with short bowel syndrome. Warner passed away during the final stages of the study’s completion; his name appears on the paper, as does a dedication in the acknowledgements. Among the patients he spent his career trying to help were premature infants with necrotising enterocolitis, a condition in which intestinal tissue dies and must be removed. These children often grow up to face exactly the liver complications WUSTL0717 is designed to prevent.
“The absence of therapies for patients with short bowel syndrome has profound implications for their long-term health,” says Colin Martin, a paediatric surgeon at WashU who treated such patients alongside Warner and is a co-author on the study. “These preclinical findings represent a crucial leap forward in our goal of developing a treatment that safeguards liver function and improves nutrient absorption, enhancing the quality of life for patients affected by short bowel syndrome.”
Whether those parcels of fat and protein, assembled in the gut lining and dispatched to the liver, turn out to be the mechanism the field has been looking for is still an open question. But it is becoming a less open one.
Source: Kim A et al., “A Gut-Restricted Liver X Receptor Agonist Ameliorates Liver Injury in Experimental Short Bowel Syndrome,” Gastroenterology, March 6, 2026. DOI: 10.1053/j.gastro.2025.12.015
Frequently Asked Questions
After major bowel resection, the gut can no longer produce normal quantities of HDL particles, which travel through the portal vein and appear to help protect the liver from inflammatory signals originating in the gut. Without that protective shield, the liver accumulates scar tissue over time a process called fibrosis that can eventually progress to liver failure. The connection between gut length and liver health is more direct, and more biochemical, than most people realise.
The HDL that matters here is made in the gut itself and travels through the portal vein a route that connects the intestine directly to the liver. Conventional HDL-boosting drugs tend to work systemically and have their own complication profiles; earlier LXR agonists, the same class as WUSTL0717, triggered dangerous liver fat accumulation when they reached the liver. The new compound’s trick is staying in the gut, which means it gets the benefit without the side effect.
The next step is testing WUSTL0717 in larger animals under conditions that more closely resemble clinical short bowel syndrome, including the use of intravenous nutrition, which itself stresses the liver. If those results hold, human trials would follow. The compound’s chemical profile it degrades quickly and doesn’t accumulate systemically is a useful starting point for regulatory assessment, but the path from mouse model to approved drug typically takes many years.
The gut-to-liver HDL pathway is likely relevant to other conditions where the intestine is compromised, including inflammatory bowel disease and the liver complications sometimes seen after bariatric surgery. The broader principle that targeting a receptor specifically in the gut can influence liver health without the toxicity of systemic drug exposure could also inform how researchers approach other gut-liver axis targets that have previously seemed too risky to pursue.
ScienceBlog.com has no paywalls, no sponsored content, and no agenda beyond getting the science right. Every story here is written to inform, not to impress an advertiser or push a point of view.
Good science journalism takes time — reading the papers, checking the claims, finding researchers who can put findings in context. We do that work because we think it matters.
If you find this site useful, consider supporting it with a donation. Even a few dollars a month helps keep the coverage independent and free for everyone.