The mice in Aaron Burberry’s lab at Case Western Reserve University kept dying. Genetically, they were identical to mice at other institutions around the country. Same breed, same mutation, same expected outcomes. But the ones in Cleveland were developing severe inflammation and dying months before their counterparts housed at the Broad Institute in Cambridge, Massachusetts, or the Jackson Laboratory in Bar Harbor, Maine. The difference, it turned out, wasn’t in their genes. It was in their guts.
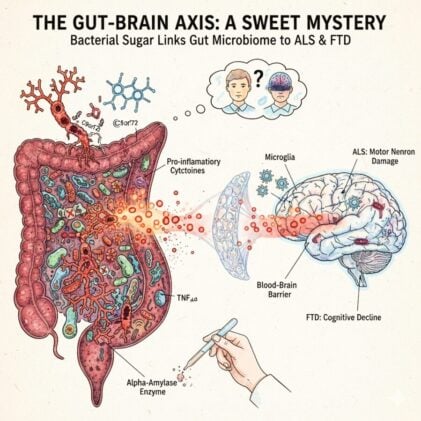
Burberry and his team have now traced the problem to something deceptively simple: a type of sugar called glycogen, produced by certain gut bacteria, that triggers an immune cascade capable of breaching the blood-brain barrier and damaging neural tissue. The discovery, published in Cell Reports, could reshape how we think about two of the most devastating neurological conditions, amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD), by pointing to the gut microbiome as a modifiable trigger for brain disease.
ALS destroys motor neurons, leading to progressive paralysis and typically death within two to five years. Up to 15 per cent of people with ALS also develop FTD, which erodes personality, behaviour and language by attacking the brain’s frontal and temporal lobes. The two conditions sit on a shared clinical spectrum, and the most common genetic cause of both is a mutation in a gene called C9ORF72, accounting for roughly 10 per cent of cases. But here’s the nagging puzzle that has haunted researchers for years: not everyone who carries the mutation gets sick. Something environmental has to be pulling the trigger.
That something, Burberry’s team now argues, is bacterial glycogen.
To crack the case, the researchers took a methodical approach. They collected faecal matter from mice housed at four different institutions and exposed it to immune cells called macrophages. Faeces from the pro-inflammatory environments (Cleveland and Harvard) provoked far more of the cytokine TNF-alpha than samples from the protective facilities. Crucially, this exaggerated response only occurred in macrophages lacking C9orf72, suggesting the gene normally acts as a brake on inflammation triggered by gut microbes.
Next came the detective work of identifying which bacteria were responsible. The team tested 18 bacterial strains individually and found 10 phylogenetically diverse species that ramped up cytokine release in a C9orf72-dependent way. Metatranscriptomic sequencing pointed the finger at a specific metabolic pathway: glycogen biosynthesis. A machine learning algorithm trained on the data classified faecal samples from pro-inflammatory versus protective environments with near-perfect accuracy, an area under the curve of 0.98. When the researchers treated the offending bacteria with alpha-amylase, an enzyme that breaks down glycogen, the inflammatory signal from four of the five key species dropped substantially. The sugar was the weapon.
Under the electron microscope, you could actually see it. Bacteria treated with alpha-amylase looked hollowed out compared with untreated controls, their intracellular density visibly reduced, consistent with the depletion of glycogen stores. And this wasn’t just any glycogen. Structural analysis revealed that the inflammatory forms had shorter sugar chains and more frequent branching than the benign varieties, a compact architecture that makes them more thermodynamically stable and, apparently, more irritating to the immune system.
“We found that harmful gut bacteria produce inflammatory forms of glycogen… and that these bacterial sugars trigger immune responses that damage the brain,” said Burberry. The finding explains a conundrum that has lingered for decades. Why do some C9ORF72 mutation carriers develop ALS or FTD while others live out their lives unaffected? The answer may depend partly on which microbes have colonised their intestines.
The most dramatic evidence came from germ-free mice. The team re-derived their C9orf72-deficient animals in completely sterile conditions, then selectively introduced different bacterial communities. Mice that remained germ-free showed only mild inflammation. Those given protective bacteria from Jackson Laboratory fared reasonably well. But mice colonised with a single species, Parabacteroides merdae, alongside otherwise benign gut flora developed a severe inflammatory syndrome: enlarged spleens, elevated monocytes, T cells infiltrating the spinal cord, and immunoglobulin leaking across the blood-brain barrier into the brain. With C9orf72 intact, these same bacterial exposures caused far less damage.
Then the team tried something that sounds almost too straightforward. They took conventionally housed C9orf72-deficient mice that were already showing signs of disease (poor motor performance, elevated blood markers, about six months old) and began daily oral doses of alpha-amylase. Over 10 weeks, every single treated mouse survived. Among the vehicle-treated controls, three of six males died. The enzyme also reduced spleen size and dampened a C9orf72-dependent inflammatory gene signature in brain microglia, essentially redirecting these immune cells away from their preoccupation with circulating bacterial debris. Alpha-amylase didn’t fix everything; motor performance didn’t improve, and blood markers stayed stubbornly elevated. But the treated animals lived, and their brains were less inflamed.
When the researchers turned to human samples, the pattern held up. They obtained faecal matter from 23 patients with ALS or FTD and 12 healthy controls from centres in the United States and Italy. Inflammatory glycogen was present in the guts of about 70 per cent of the patients, but only a third of healthy individuals. Across the ALS/FTD group, the alpha-amylase-sensitive signal was statistically significant. Among healthy controls, it wasn’t. Eight of nine samples collected within the first 18 months of an ALS diagnosis contained inflammatory glycogen, hinting that the sugar might be especially relevant early in disease.
The findings are suggestive rather than definitive, as the human cohort was small and the study couldn’t determine whether inflammatory glycogen preceded disease onset. Burberry acknowledges that bacterial glycogen is likely one environmental factor among many that interacts with predisposing genotypes. But the therapeutic implications are tantalising. Reducing harmful sugars in the gut “improved brain health and extended lifespan” in the mouse model, according to Rodriguez-Palacios. Clinical trials could begin within a year, Burberry indicated, to test whether degrading glycogen in the guts of ALS and FTD patients might slow disease progression.
There’s a broader lesson here, too. We tend to think of neurodegeneration as a problem that starts and ends in the brain. But the gut harbours an estimated 100 trillion microorganisms interfaced by 70 to 80 per cent of the body’s immune cells. What if, for some people carrying the genetic dice for ALS or FTD, the critical moment isn’t a failure inside a motor neuron at all, but a particular bacterium in the colon switching on a glycogen pathway? It’s a disconcerting thought, but also, perhaps, a hopeful one. You can’t change your genes. But you might, eventually, be able to change what your gut bacteria are cooking up.
Study link: https://www.cell.com/cell-reports/fulltext/S2211-1247(25)01678-X
ScienceBlog.com has no paywalls, no sponsored content, and no agenda beyond getting the science right. Every story here is written to inform, not to impress an advertiser or push a point of view.
Good science journalism takes time — reading the papers, checking the claims, finding researchers who can put findings in context. We do that work because we think it matters.
If you find this site useful, consider supporting it with a donation. Even a few dollars a month helps keep the coverage independent and free for everyone.