Key Takeaways
- Central vision loss results from the death of cone photoreceptors, which traditional treatments cannot halt.
- A team at the Institute of Molecular and Clinical Ophthalmology Basel screened nearly 2,700 compounds on human retinal organoids, identifying casein kinase 1 as a potential protective target.
- Two compounds, CS-KI-1 and CS-KI-2, showed promise in protecting cones against death, while several HDAC inhibitors proved harmful to them.
- The research highlights the potential toxicity of HDAC inhibitors in cancer treatments for patients with retinal conditions, suggesting a need for further investigation.
- The newly developed database includes 2,707 compounds tested for cone survival, aiding future drug development and toxicity assessments.
Central vision loss is, in a very particular sense, a slow erasure. The cells responsible for reading, for recognising faces, for the whole rich foreground of human sight are called cone photoreceptors. And in conditions like age-related macular degeneration or advanced retinitis pigmentosa, they die in their millions over years, leaving behind a spreading blind spot that no existing medicine can stop. Not slow the progression. Not delay. Stop. That goal has eluded ophthalmology for decades, largely because testing potential treatments in human retinal tissue was, until recently, essentially impossible.
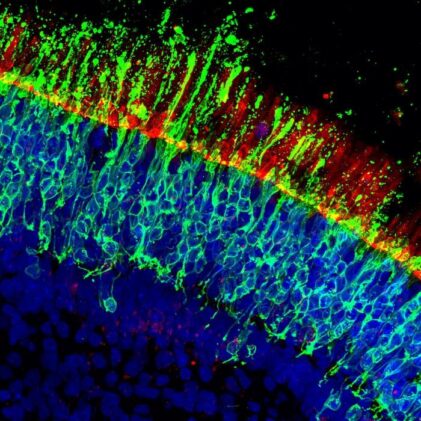
A team at the Institute of Molecular and Clinical Ophthalmology Basel has now run what may be the largest human retinal drug screen ever attempted, growing roughly 20,000 miniature retinas in the laboratory and testing nearly 2,700 compounds against them in search of anything that might keep cones alive. The results, published in Neuron, point toward an unexpected target: a kinase enzyme called casein kinase 1.
The scale of the operation is worth dwelling on, because it’s what made the findings possible. Botond Roska’s group, working with first authors Stefan Spirig and Alvaro Herrero-Navarro, grew human retinal organoids from induced pluripotent stem cells over 30 weeks until each tiny sphere contained five layers of organised retinal tissue, cones included. They then labelled the cones with a green fluorescent protein delivered via viral vector, which meant you could watch individual cone deaths in real time under a confocal microscope, tracking how many survived across a full 96-well plate of organoids in a single automated scan. Specificity of labelling: 97%. Efficacy: around 64%. Good enough for a systematic screen.
Cones are among the most metabolically demanding cells in the body, packed into the macula with almost no redundancy and no real ability to regenerate once lost. In many diseases, their death is a secondary event triggered by failure elsewhere in the retina, which means by the time cones are dying, the underlying problem has often been going on for years. Testing drugs that might slow or stop cone death has historically required animal models that don’t translate well to humans, which is part of why this organoid-based screen, using actual human retinal tissue, is a bit of a step change for the field.
Casein kinase 1 is a family of enzymes involved in regulating a wide range of cellular processes, including circadian rhythms, cell division, and the Wnt signaling pathway that governs cell growth and fate. Different members of the family (CSNK1G1, CSNK1G3, and others) are found throughout the body, which is part of what makes it a tricky therapeutic target. The researchers found that knocking down specific CK1 family members only in cone photoreceptors was enough to improve their survival, suggesting the relevant action is happening inside the cones themselves rather than somewhere else in the retina.
Possibly, though it’s early. The compounds identified here haven’t been through human safety testing, and CK1 is expressed widely enough across the body that a systemic drug could have unintended effects. Intravitreal injection (directly into the eye) is already standard for macular degeneration treatments like anti-VEGF drugs, so a locally delivered CK1 inhibitor might sidestep some systemic concerns. The researchers have filed a patent on aspects of the findings, which at minimum suggests confidence the target is real.
HDAC inhibitors are being actively tested as cancer treatments, and some are already approved. The screen found that broad HDAC I/II inhibitors are among the most reliably toxic compounds to cone photoreceptors, in a dose-dependent way. That raises a practical clinical question: are cancer patients on these drugs at elevated risk of retinal damage? The public database from this study gives toxicologists and clinicians a specific, human-tissue-derived reference for checking which HDAC inhibitors look most problematic for cones specifically.
To actually kill the cones, the researchers deprived organoids of glucose. This isn’t arbitrary: there’s a running hypothesis in the field that cones in retinitis pigmentosa starve to death once the rods (which ordinarily supply them metabolic support) are gone. Within 7 days of low-glucose medium, roughly 40% of cones had died. A manageable killing window for a compound screen.
What the screen turned up fell into two camps, and honestly, the bad news is nearly as useful as the good. A whole class of compounds that damages cones, perhaps quite badly. Histone deacetylase inhibitors, HDAC I/II inhibitors specifically, turned up again and again as cone-killers: 19 of the 146 most damaging compounds in the primary screen were HDAC I/II inhibitors, and the probability that this was random chance came out at less than 0.0001. The broader the range of HDAC targets a compound hit, the worse the cone damage. These drugs are actively being investigated as cancer treatments. The implication, somewhat inconvenient for oncology, is that retinal toxicity may be an underappreciated side effect in patients who happen to have pre-existing eye conditions.
The protective side of the ledger took more unpicking. Several compounds looked promising in the 7-day window but fell apart at 14 days: inhibitors of a heat-shock protein called HSP90AA1 saved cones in the short term and then turned on them. Not a great quality in a potential therapy. Two other kinase inhibitors, which the team labelled CS-KI-1 and CS-KI-2, were different.
CS-KI-1 and CS-KI-2 protected cones at both 7 and 14 days, and also protected rod photoreceptors, which was something the researchers weren’t even initially screening for. But identifying two useful compounds is only half the work; the harder question is why they work, because knowing the target is what lets you design better drugs. The original annotation on both compounds pointed to targets (mTOR for CS-KI-1, PDGFR for CS-KI-2) that turned out to be almost certainly wrong. The researchers used a clever trick: they found chemical analogs of each compound that were structurally very similar but didn’t protect cones, then ran kinase profiling across 350 human kinases to see what each active compound inhibited that its inactive twin did not. For CS-KI-1, the differentiating target was CSNK1G1, a member of the casein kinase 1 family. For CS-KI-2, it was MAPK11. Three independent CK1 inhibitors were then tested, and all three protected cones significantly. To confirm that the effect was happening inside the cones themselves rather than through some other cell type, the team used short hairpin RNAs to knock down CSNK1G1 and another CK1 family member, CSNK1G3, specifically in cones. Cone survival improved. The protection is, it seems, intrinsic to the photoreceptors.
The glucose starvation model could, in principle, be a quirk, so the team also induced cone death via oxidative stress (hydrogen peroxide this time) and found that CK1 inhibitors protected cones there too. Different insult, similar rescue.
In vivo data came from rd10 mice, a standard model of retinitis pigmentosa in which photoreceptors progressively degenerate. Injections of CS-KI-1, CS-KI-2 and CK1-I-1 directly into the eye, weekly from postnatal day 14 to 35, measurably preserved retinal thickness relative to vehicle-injected control eyes. Not dramatic preservation, but statistically significant, and in a degenerating retina, measurable preservation is something.
There are obvious caveats. Organoids aren’t retinas, and mice aren’t people. The casein kinase 1 family has many members spread across many tissue types, which means systemic CK1 inhibition might cause problems elsewhere in the body. The compounds here haven’t been through any toxicology, or pharmacokinetics, or clinical trials. The road from “interesting target in lab-grown retinas” to “approved therapy for macular degeneration” is long and lined with failures.
What’s perhaps more immediately useful is the database the team is making publicly available: all 2,707 compounds, their molecular targets, and their effects on cone survival, searchable at ConeTargetedCompoundScreen.iob.ch. That resource matters both for people developing potential neuroprotective drugs and for people assessing retinal toxicity risk in existing drug pipelines, the HDAC inhibitor finding being a case in point. A screen this size, in human tissue, didn’t really exist before. It does now.
Casein kinase 1 had no particular profile in retinal biology before this work. It does now, and the question the field will likely spend the next several years trying to answer is why blocking it keeps photoreceptors from dying, and whether that insight can be turned into something a patient might actually take.
DOI / Source: https://www.cell.com/neuron/fulltext/S0896-6273(26)00129-7
ScienceBlog.com has no paywalls, no sponsored content, and no agenda beyond getting the science right. Every story here is written to inform, not to impress an advertiser or push a point of view.
Good science journalism takes time — reading the papers, checking the claims, finding researchers who can put findings in context. We do that work because we think it matters.
If you find this site useful, consider supporting it with a donation. Even a few dollars a month helps keep the coverage independent and free for everyone.